近日,吉林大学超分子结构与材料国家重点实验室张越涛教授课题组,合成了一种分子内三官能FLP催化剂,催化γ-methyl-α-methylene-γ-butyrolactone (MMBL)合成了完全由一种丙烯酸酯类单体构成的、无端基的、真正的环状聚合物c-PMMBL。该成果以标题为“Recyclable cyclic bio-based acrylic polymer via pairwise monomer enchainment by a trifunctional Lewis pair”发表在最新一期Nature Chemistry上(DOI:10.1038/s41557-022-01097-7)。该工作的第一作者和通讯作者分别是吉林大学化学学院超分子结构与材料国家重点实验室的宋艳娇博士和张越涛教授。
环状聚合物是一类没有链端结构的特殊拓扑类型的聚合物。其一般具有区别于传统线型聚合物的性质,如更低的黏度、更高的热稳定性等。然而,受限于合成方法少、难度大等原因,使得人们对环状聚合物性能的了解依然有限。因此当前研究的热点和难点是发展环状聚合物的高效、可控合成方法。
目前获得环状聚合物的合成策略主要分为两种:关环策略(RCP)和扩环策略(REP)。前者是将单分子或双分子聚合物链的α端和ω端进行偶联得到环状聚合物,这种方法需要对聚合物链端进行官能化,而且偶联成环需要在极稀释条件下进行;后者虽然可以在高浓度条件下合成环状聚合物,但由于缺乏普适性,一般需要为每种单体单独设计催化剂/引发剂。
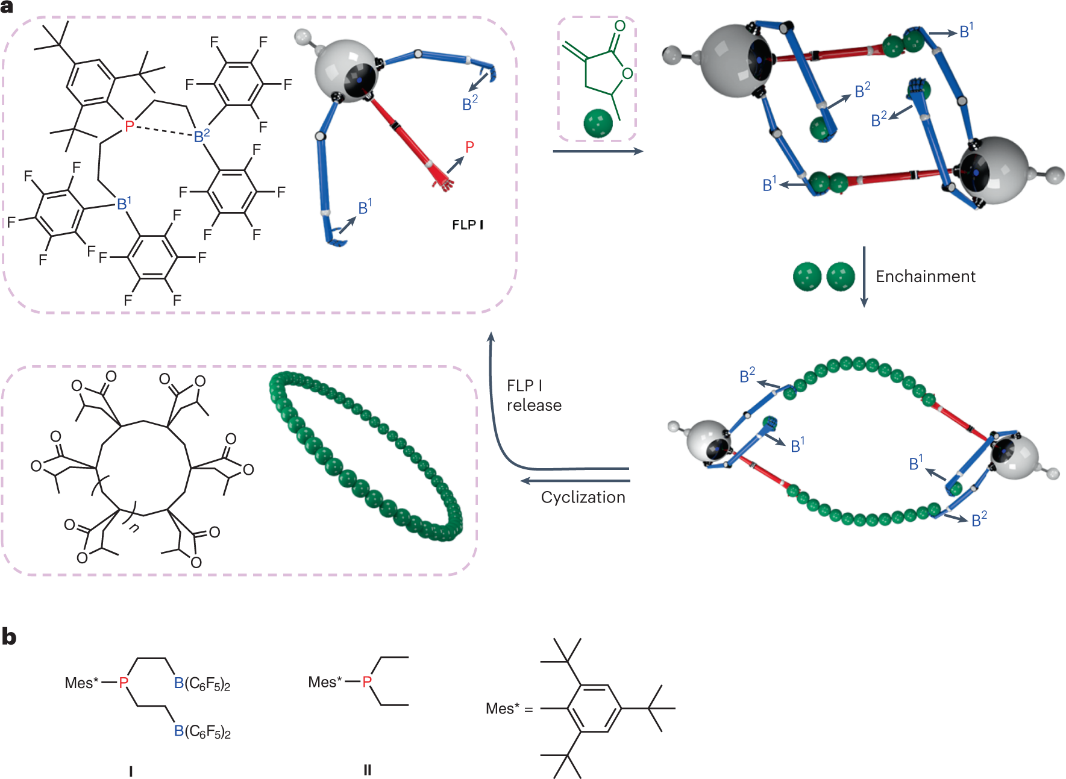
图1 a, c-PMMBL形成示意图。FLP I与单体反应形成环状二聚体模板,然后成对的单体嵌入以扩环,最终使催化剂I环化和再生。酸性B位点为蓝色,标记为B1和B2,碱性P位点为红色,单体为绿色。b,本工作中使用的三官能催化剂B-P-B(I)和单官能P(II)的结构。
通过FLP I 催化 MMBL 聚合结果来看(图1a),该催化剂具有较高的活性,几乎可以实现100-1200当量MMBL的完全转化。
作者首先对聚合物的环状拓扑结构进行了表征,通过MALDI-TOF可以看出(图2b)图中具有两组峰,意味着似乎存在两种不同的链引发/链终止途径,但分析发现二者均为不含端基的环状结构,区别仅在于聚合物链中单体数量是奇数还是偶数。在证明了聚合物的环状拓扑结构的同时,上述特殊的结果也激起了作者对机理进行更深入研究的兴趣。作者通过制备了与环状聚合物分子量近似的线型聚合物,比较二者的特性粘度[η]I/[η]linear ≈ 0.7(图2e)。进一步确定了PMMBL的环状拓扑结构。

图2 a, 表中显示了室温(~25℃)下由MMBL和FLP I在DCM中制备的PMMBL的选定聚合数据 ,[M]0 =?0.936?M. a通过1H NMR光谱测定单体转化率。b使用光散射检测器通过GPC测量绝对重均分子量(Mw)。cMn 由Mw/?计算得出. d? = Mw/Mn. b, 用FLP I.制备的MMBL低聚物的MALDI-TOF MS谱,c,d,b中分子离子的主要(c)和次要(d)系列的m/z与2MMBL重复单元数量的关系图。e,l-PMMBL和c-PMMBL的特性粘度的比较,为用IAP–Al(C6F5)3 制备的l-PMMBL和用I制备的c-PMMBL的Mark–Houwink图。f, c-PMMBL50-ran-PVMBL50-g-PEG2000的TEM图像。
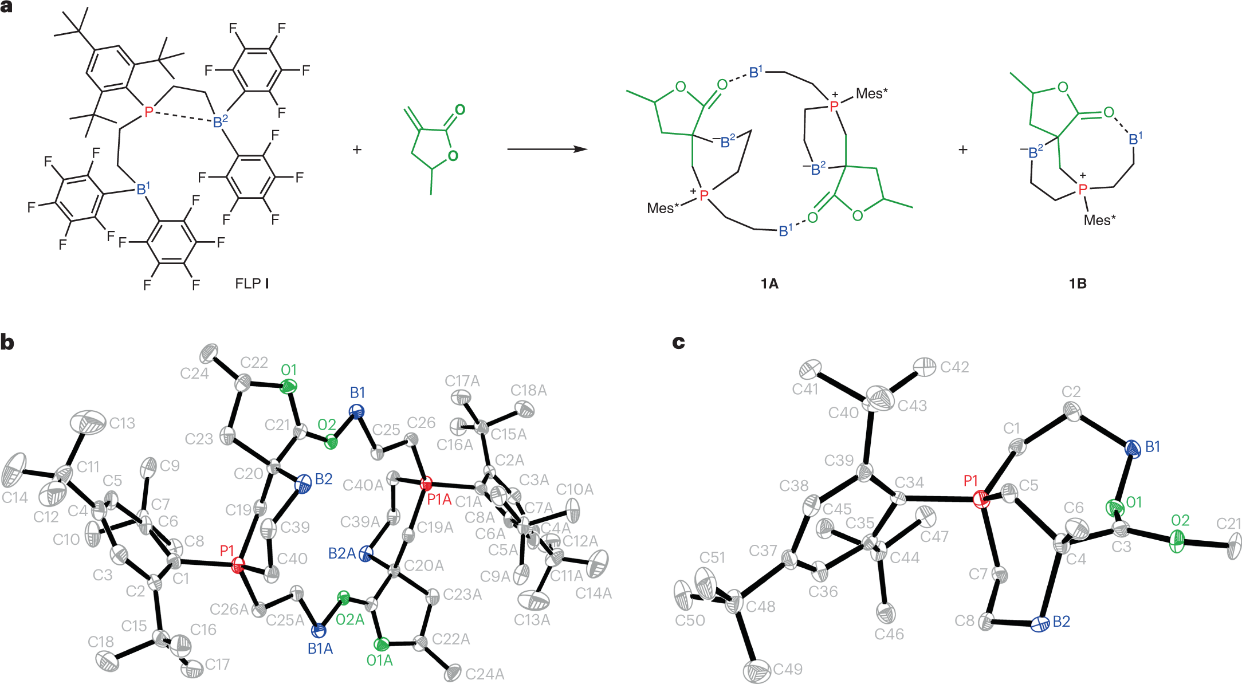

图4 a,b,室温下使用1A (a)和 1A和 1B (b)在DCM中的混合物进行MMBL聚合的零级动力学图。插图:ln kapp 与 ln?[P]total 图 总共从两组聚合动力学中提取。 [P]total =?2?×?[1A]?+?[1B],表示系统中P位点的总浓度。c,分别从起始的双分子环1A和单分子环1B模板到催化剂-单体络合物D和M的基本步骤,以及它们对应的单体嵌段过渡状态。还显示了1A和1B所提出的RDSs的相应动力学方程。

图5 c-PMMBL合成中的关键基本步骤,包括通过将催化剂以相同的化学计量加入到MMBL中形成环状模板1A和1B,环状模板1A、1B中的后续单体配位/活化和插入事件以及双催化剂过渡状态,通过在双催化剂过渡状态和环闭合的两个LP催化剂位点处同时重复成对单体嵌段进行环膨胀,以产生具有奇数和偶数MMBL单元的环状聚合物并再生催化剂。
作者还对该单体的普适性做了研究,实现了MMBL与3-methylene-5-vinyldihydrofuran-2(3H)-one (VMBL)的随机共聚(MMBL/VMBL/I = 50:50:1)。不仅如此,还通过点击反应对VMBL的双键进行后官能化,优于侧链的引入可以在TEM下观测到该环状接枝共聚物的图像(图2f),从而以更直观的方式表征了该类聚合物的环状拓扑结构。
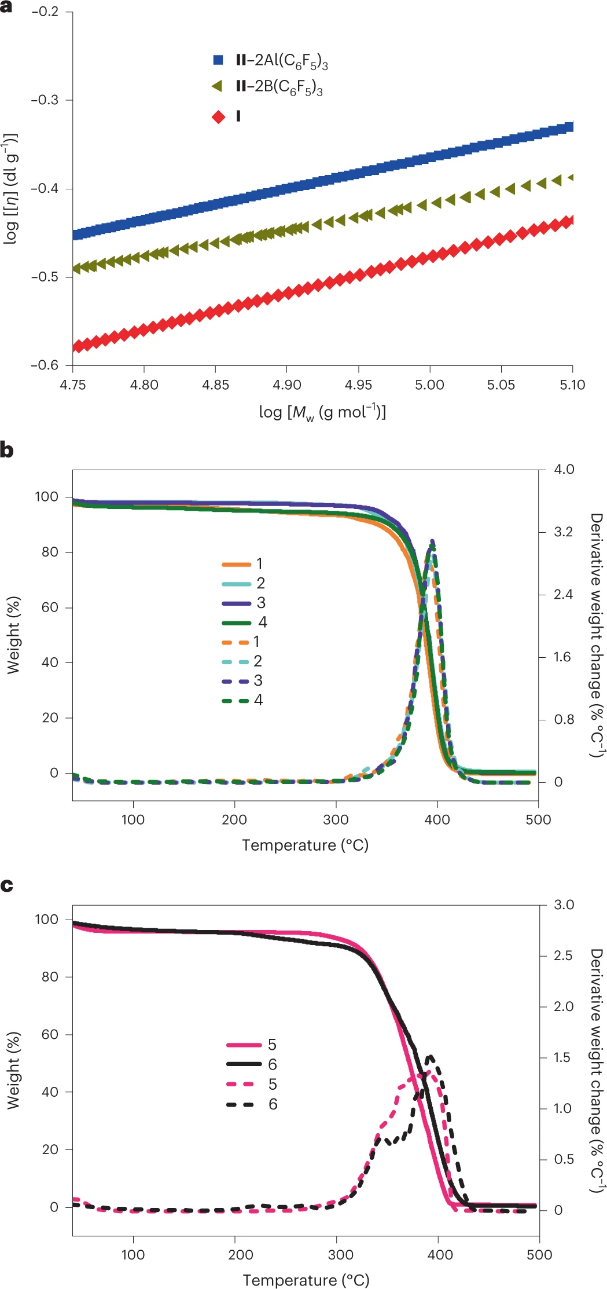
图6 a,不同拓扑结构的PMMBL的特性粘度比较,如Mark–Houwink图所示:由FLP I产生的c-PMMBL,由分子间 II–2B(C6F5)3 产生的环状和线性PMMBL混合物,以及由分子间LPII–2Al(C6F5)3制备的l-PMMBL。b,具有四种不同MWs的c-PMMBL样品的TGA(实线)和DTG(虚线)曲线叠加:sample 1, Mn =?93.1?kg?mol–1, ? =?1.31; sample 2, Mn =?109?kg?mol–1, ? =?1.21; sample 3, Mn =?176?kg?mol–1, ? =?1.41; sample 4, Mn =?403?kg?mol–1, ? =?1.88. c,两种不同MWs的l-PMMBL样品的TGA(实线)和DTG(虚线)曲线叠加图:sample 5, Mn =?289?kg?mol–1, ? =?1.82; sample 6, Mn =?427?kg?mol–1, ? =?1.77.
原文链接:https://www.nature.com/articles/s41557-022-01097-7
- 华理胡爱国教授、王贵友副教授团队 Macromolecules:通过纳米限域下的Suzuki交叉偶联缩聚合成窄分布的环状聚合物 2023-12-02
- 南华大学魏华/喻翠云团队ACS Macro Lett.封面:紫外光诱导环状聚合物载体的环状-线形拓扑结构变化用于胞内增强药物递送 2023-08-16
- 吉林大学吴宗铨教授团队 Angew: 手性环状螺旋聚合物的可控合成及其圆偏振发光 2022-05-10
- 哈工程刘天亿、复旦大学孔彪 ACS Nano:非对称聚合物半导体纳米机器人的可控合成与感染皮肤创面治疗 2026-04-16
- 长春应化所陶友华研究员团队 JACS:阴离子结合催化实现聚(1,3-二氧戊环)的可控合成 2026-03-27
- 华东理工刘润辉教授课题组 Angew VIP:阳离子催化剂实现氨基酸聚合物的超快速可控合成及高效解聚 2025-11-05
- 四川大学朱剑波教授团队 Angew:利用催化剂对映体过量值实现聚3-羟基丁酸酯的精准立体调控 2026-07-28
