聚酯材料在当今的社会生活中占据着重要的地位,其应用范围涵盖了织物、餐饮器具、农用薄膜和医疗器械等领域,全球年消耗量超过了7000万吨。目前采用商品化的二元羧酸和二元醇进行熔融缩合聚合是大规模工业生产聚酯最经济的途径。聚酯工业中普遍采用金属催化剂来催化酯交换反应,脱除过量的二元醇来提高产物分子量。然而,这些具有较高生物毒性的金属催化剂会残留在聚酯基体中,对人体和自然环境造成危害。早在1929年,高分子学科的先驱Wallace H. Carothers就研究了二元羧酸与二元醇可在羧酸单体自催化下进行酯化反应获得相应聚酯,不需要外加任何催化剂。然而产物分子量仅有2~5 kDa,性能太差而无法应用。近一个世纪后,在催化剂残留对健康及环境的影响越来越受到重视的背景下,重新研究自催化酯化缩聚难以获得高分子量聚酯的真实原因,探索提高自催化缩聚效率的新机理和新途径,彻底解决聚酯中催化剂残留的问题,具有重大理论意义和应用价值,但也面临巨大挑战。
热力学因素,即酯化反应的低平衡常数和高熔体黏度下排除副产物水的困难,被普遍认为是导致自催化方法无法获得高分子量聚酯的原因。然而,工业上广泛使用的酯交换法可在金属催化剂催化下获得高分子量的聚酯产物,其中酯交换反应的平衡常数(< 1)甚至比酯化反应(~4)更小、副产物(如乙二醇)比水更难排除,这就说明了热力学因素应当不是自催化法无法获得高分子量产物的原因。在动力学上,获得高分子量聚酯还要求达到醇酸基团等摩尔比这样的苛刻条件。而自催化反应为三级反应,即反应速率与羟基浓度的一次方和羧基浓度的二次方成正比。实际反应过程中,不仅由于基团浓度下降导致后期聚合速率过慢,而且醇酸基团常常偏离等摩尔比,导致聚酯的分子链无法增长,这也正是传统酯交换法所解决的主要问题。因而,动力学因素应当是限制自催化下聚酯产物分子量提高的关键。

基于上述的新认识,浙江大学高分子科学与工程学系朱蔚璞副教授团队提出了一种新型的无催化剂缩聚(CFP)机理,该机理采用了一类能够形成五元环或者六元环酸酐的二元羧酸作为单体(图1)。
过量的此类二元酸与伯二元醇酯化形成羧基封端预聚物后,发生如下的串联反应:
-
(1)羧基封端预聚物发生可逆的分子内和分子间质子转移,分别形成质子化的羧基阳离子和羧基两性离子;
-
(2)分子链末端的羧基两性离子“回咬”生成环状酸酐,再生出羟基基团;
-
(3)再生的羟基和羧基阳离子再次进行酯化反应并脱除副产物水,使得分子链发生增长。
通过上述串联反应,使得体系中的醇酸基团比不断趋近于1:1,产物分子量增长呈现出独特的“加速”模式。从而在与传统工艺相近的时间内,通过熔融缩聚获得了一系列的高分子量无催化剂聚酯,包括聚丁二酸丁二醇酯(PBS)、聚丁二酸乙二醇酯(PES)、聚(丁二酸丁二醇酯-共-己二酸丁二醇酯) (PBSA)和聚(丁二酸乙二醇酯-共-对苯二甲酸乙二醇酯) (PEST)等已商业化的可降解聚酯。由此,证实了传统认知的热力学困难并非是导致自催化法中聚酯分子量难以提高的主要原因。目前该研究成果以题为Catalyst-free synthesis of polyesters via conventional melt polycondensation的研究论文发表在Materials Today上,并被选为封面论文。

图1. CFP法合成聚酯的机理
作者首先对反应机理进行了研究。使用摩尔比过量10%的丁二酸(1a)与1,4-丁二醇(2a)在无外加催化剂、200°C的条件进行酯化反应,获得了羧基封端的PBS预聚物。1H NMR谱图(图2a)证实此PBS预聚物中的丁二酸单元与丁二醇单元之比(记为R/R’)为1.11:1,说明其的确为羧基封端。该PBS预聚物对应的数均分子量(Mn)和黏均分子量(Mη)分别为1.2和1.3 kDa。随后,为了确认是否可以通过PBS预聚物链末端的“回咬”产生酸酐,将此预聚物置于密闭烧瓶中并在240°C、常压下加热至反应达到平衡。结果发现,在加热后的混合物的1H NMR图谱中(图2a)清晰地出现了大量丁二酸酐(3a)的峰,并且相对于二元醇单元,其含量(6%)与剩余的丁二酸单元的量(105%)之和刚好与加热前的丁二酸单元的量(111%)吻合。与此形成对比的是,在加热前的PBS预聚物中几乎检测不到丁二酸酐的存在。该结果使得最终产物的分子量显著增大了3倍,达到了Mn = 3.6 kDa和Mη = 3.5 kDa。而且,PBS预聚物的MALDI-TOF-MS谱图由三个系列的分子离子峰构成(图2b),其中最大峰及其邻近的小峰归属于两端均为羧基的聚酯链(P1)的钠离子和钾离子加成物,而第三系列的小峰是一端为羧基一端为羟基的聚酯链(P2)的钠离子加成物。作者发现,P2的持续性产生是聚酯分子量能够增长的关键。其证据为在同等的反应条件下,使用丁二酸与摩尔比过量10%的1,4-丁二醇进行反应时,其体系中并未探测到P2的存在。并且,在难以形成酸酐的己二酸(1c)参与的体系中观察到分子链主要以P1型为主,而P2型只有极少量。上述结果证明分子链端生成环酸酐的能力是聚酯分子量能否增长的关键。

图2. 反应机理研究。(a)PBS预聚物(上)及其在240°C密闭加热至反应平衡后(下)的1H NMR谱图对比; (b) MALDI-TOF-MS谱图显示PBS预聚物由两端均为羧基的分子链(P1)与一端羧基一端羟基的分子链(P2)构成
为了提高生成酸酐反应的反应程度,作者将实验条件优化为高温和高真空(240 °C, < 100 Pa)。在该条件下,PBS预聚物中的醇酸单元比(R/R’)在5 h内从初始的1.1:1降至接近1:1,最终产物的黏均分子量达到了64.7 kDa (表1,entry 1)。这也说明了通过二元羧酸的自催化来获得高分子量聚酯是完全可行的。然而,将聚己二酸丁二醇酯(PBA)预聚物按照同样的条件操作后,发现产物的醇酸单元比R/R’并未产生明显变化,最终的数均分子量也只有3.9 kDa (表1, entry 3)。作者进一步对多种二元羧酸和二元醇单体的适用性进行了验证。例如,将可形成环酸酐的戊二酸(1b)、(±)-甲基丁二酸(1f)、2,2-二甲基丁二酸(1g)、(±)-苯基丁二酸(1h)和(±)-巯基丁二酸(1i)和二甘醇酸(1j)与1,4-丁二醇反应可获得高分子量产物 (表1, entry 2、6~10)。而更长碳链的难形成酸酐的二元羧酸如庚二酸(1d)和癸二酸(1e),最终产物分子量如预期的较低(表1, entry 4、5)。然而发现了三种例外的情形:当采用马来酸(1k)、邻苯二甲酸(1l)和(+)-樟脑酸(1m)具有共轭或者双环结构的短链二元酸时,却仅能获得低聚物 (表1, entries 11~13)。以上这些结果表明聚酯产物的分子量应该与酸酐形成反应和酯化反应的相对速率有关。因而,对于两端都是伯羟基的二元醇,可获得高分子量产物(表1, entries 14~16),而使用具有仲羟基或者叔羟基的二元醇反应后也仅能获得低聚物(表1, entries 17、18)。
表1. 采用多种二元酸与二元醇单体合成无催化剂聚酯
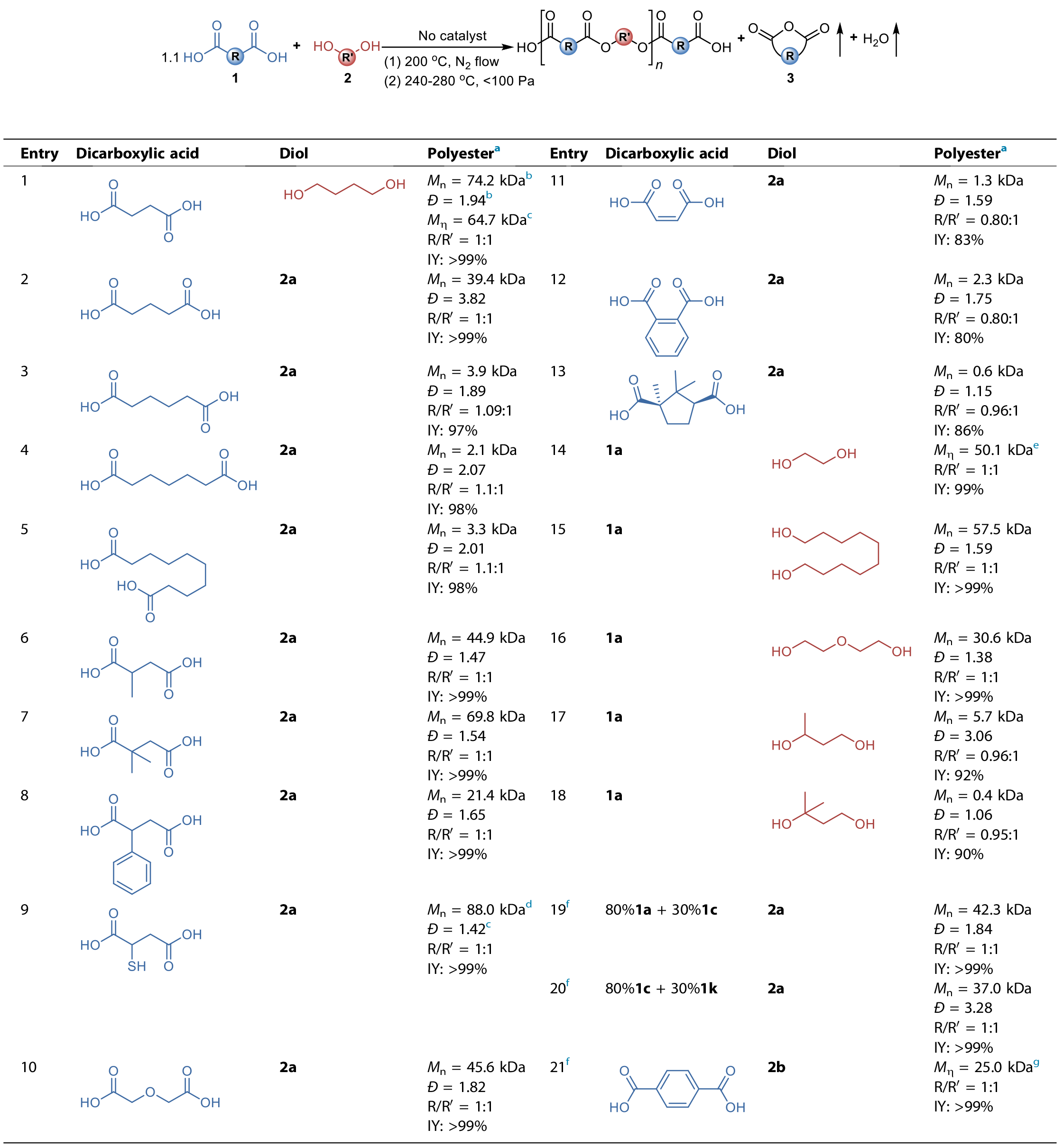
作者还通过密度泛函理论(DFT,图3a)计算出了酸酐形成反应的理论反应热(ΔΔG?)和理论反应活化能(ΔΔG?max),并分别对实验测定的反应平衡常数K和反应速率常数k1的对数作图,结果发现它们之间具有高度的线性相关性(图3b和3c)。能够获得高分子量聚酯的体系中,其适宜的ΔΔG?和ΔΔG?max区间分别为4.61~10.97 kcal·mol-1和45.74~55.29 kcal·mol-1。而不能获得高分子量的体系,则均在此区间外。由此,通过计算上述理论值是否落于合适区间,就能够预测其他类型的二元羧酸的适用性。其深层原因在于酸酐形成反应的速率能够与再酯化反应的速率相匹配,相对过高或过低的酸酐形成速率均无法获得高分子量产物。基于以上认识,作者还将无催化剂缩聚法推广到难以形成或无法形成酸酐的二元羧酸单体,通过与可形成酸酐的二元酸单体的共缩聚,使得上述两步反应的相对速率能够匹配,成功地获得了高分子量的PBSA、聚(己二酸丁二醇酯-共-马来酸丁二醇酯)和PEST(表1,entries 19~21)。

图3. DFT研究及其预测性。(a)DFT计算的酸酐形成过程的自由能变化; (b)理论计算的ΔΔG?与实验测定的lnK呈高度线性相关;(c)理论计算的ΔΔG?max与实验测定的lnk1呈高度线性相关
最后,作者也评估了所合成的无催化聚酯能否达到商业产品的标准。图4a中展示了合成的无催化剂PBS与三种商业牌号的PBS,即巴斯夫H1200、三菱GS pla AZ91TN和昭和Bionolle 1001MD黏均分子量(Mη)和数均分子量(Mn)对比。结果显示,无催化剂PBS的这两项参数超过了巴斯夫和三菱的商品PBS。而昭和的PBS使用具有生物毒性的二异氰酸酯进行扩链,虽然达到了最高的Mη = 116.0 kDa,但应用受到限制。图4b显示,无催化剂PBS在这4种样品中展示出最高的拉伸强度(49.22 ± 5.61 MPa)和较好的断裂伸长率(32.63 ± 5.87%)。作者还将无催化剂PBS体系和醇过量的酯交换PBS体系的聚合动力学进行对比(图4b)。传统的醇过量的酯交换体系,其羟基消耗速率(-d[OH]/dt)与羟基浓度([OH])遵循二级关系,由此产物的分子量正比于反应时间:Mt = kt+M0。而无催化剂聚酯体系中,羟基消耗速率(-d[COOH]/dt)与羧基浓度([COOH])是一种混合级数关系,产物的分子量与反应时间的关系为:Mt = C(ekt-1)+M0 (C、k为常数),因而分子量增长呈现出独特的“加速”模式。虽然在起始时无催化剂聚酯的分子量增长速率低于Sb2O3和SnCl2催化的两个体系,然而在聚合后期(> 4.5 h)无催化剂聚酯的分子量甚至超过了SnCl2催化的体系。最后,无催化剂体系中产生的酸酐也可以将其回收并参与到下一个聚合循环中(如图4d)。因此,通过这样的一种循环就可以实现原子的高效利用,并且可避免除水以外的副产物的产生,十分符合“绿色化学”的原则。此外,采用小鼠 MC3T3-E1 前成骨细胞和 L929 成纤维细胞对无催化剂聚酯如PBS、PES、聚癸二酸丁二醇酯(PDS)、PBSA、PEST 和聚戊二酸丁二醇酯(PBG)进行的细胞毒性试验,其细胞存活率分别在 94.2-102.4% 和 93.7-99.3% 的范围内,证实了无催化剂聚酯的良好生物相容性。

图4. PBS的比较与酸酐回收。(a)无催化剂PBS与三种商品的特性黏数([η])和分子量对比; (b)无催化剂PBS与三种商品PBS的拉伸性能对比; (c)无催化剂PBS与传统醇过量的金属催化剂(Sb2O3和SnCl2)催化的PBS的聚合动力学对比; (d)聚酯的无催化剂熔融缩聚和酸酐的回收循环示意图
综上,作者基于对二元羧酸自催化下无法获得高分子量聚酯的原因新认识,即并非是传统认知的热力学因素(酯化反应的低平衡常数和高黏度体系中脱除副产物水的困难)导致的,而是由于醇酸单元比在动力学上偏离1:1所引起的,提出了一种合成高分子量无催化剂聚酯的CFP机理。其中采用了一级二元醇和过量的可形成环酸酐的二元羧酸作为单体。在传统的两步熔融缩聚工艺下,首先获得一种羧基封端的预聚物,随后通过三步串联的基元反应:质子转移、酸酐形成和再次酯化进行分子量的增长,该机理也通过实验数据和计算机理论计算获得充分证实。而且,这种CFP法可拓展至无法形成酸酐的二元酸单体,如对苯二甲酸。通过与可形成酸酐的二元酸的共缩聚,就可以获得一系列的高分子量共聚酯。该方法不仅能够生产与现有商业产品相当的无催化剂聚酯,而且彻底避免了催化剂残留带来的负面效应,十分有望应用在具有高安全性要求的领域(如医疗器械、餐饮器具),并且在不增加成本的情况下更新当前的聚酯工艺。
该论文的第一作者和通讯作者分别为浙江大学高分子系2021届博士毕业生蔡秋泉(现就职于化学与精细化工广东省实验室,任助理研究员)和朱蔚璞副教授,浙江大学高分子系凌君教授、博士毕业生白天闻、博士生张洪杰,以及浙江大学材料学院姚旭霞博士为该论文的共同作者。该研究受到了国家自然科学基金和浙江省杰出青年基金的资助。
论文信息:Qiuquan Cai, Tianwen Bai, Hongjie Zhang, Xuxia Yao, Jun Ling, and Weipu Zhu*. Catalyst-free synthesis of polyesters via conventional melt polycondensation. Mater. Today, 2021.
https://doi.org/10.1016/j.mattod.2021.07.024
- 苏州大学王召教授团队 Angew:机械化学协同氢键催化开环聚合制备高分子量聚己内酯 2026-03-06
- 长春应化所陈学思/庞烜团队 Angew:基于电子效应调控的双功能铁催化剂催化环氧化物与环状酸酐可控共聚-合成高分子量聚酯材料 2026-03-05
- 长春应化所陈学思/庞烜/胡晨阳 Nat. Commun.:基于副反应抑制策略催化酸敏感型环氧化物与环状酸酐可控共聚-合成高分子量、可化学回收聚酯 2026-02-16
- 天津大学王彬教授/长春理工大学史新翠副教授 Adv. Mater.:三嵌段热塑性聚酯弹性体构筑的耐低温生物相容热熔胶 2026-06-23
- 天科大赵倩-李盛华课题组《Adv. Mater.》:激发依赖余辉复合材料开启6D动态加密新时代 2026-06-07
- 南昌大学姚昌广课题组 Macromolecules:通过二酮与二醇的催化转移氢化-脱氢聚合制备可循环聚酯 2026-05-27
- 浙江大学朱蔚璞教授 Adv. Mater.:废旧PET无催化剂无溶剂升级回收制备可生物降解塑料 2024-09-03
