相较于传统的、高污染且能耗高的蒽醌法,光催化二电子氧还原反应(2e? @ORR)被认为是合成过氧化氢(H2O2)的环保且高效途径。在此背景下,聚合物氮化碳(g-C3N4)因其低成本、高热稳定性和可调结构而受到广泛关注,成为2e? @ORR催化的有希望的候选材料。然而,原始g-C3N4的带隙为2.7 eV,仅允许吸收460 nm以下的光。为扩展g-C3N4的光吸收范围,开发了几种方法,如几何结构工程、供体-受体共聚合及缺陷形成。尽管这些方法扩大了光响应范围,但在扩展到可见光或近红外区域时,半导体带隙的缩小增强了光生电子与孔之间的库仑相互作用,从而降低了光生载流子的解离效率。光生载流子的解离效率(DE)可以用公式DE=e-Ec/kBT描述,其中DE代表解离效率,EC为激子库仑结合能,kB为玻尔兹曼常数。随着反向弗伦克激子的库仑力增加,解离效率下降,反向复合的概率上升,导致光生载流子分离效率显著降低,光催化性能下降。
先前的研究表明,碳掺杂g-C3N4能有效缓解上述问题,改变光催化材料的电子结构,优化带隙宽度,以促进更广谱光子的吸收,使g-C3N4能够在低能光激发下产生光激发电子和孔。此外,碳掺杂引入的额外能级能够捕获光激发电子,从而降低电子-孔对的复合率,增强激子分离。然而,之前的研究中,非晶态碳作为残留非晶碳被掺入g-C3N4,导致不规则掺杂位点和潜在的结构不稳定。纯碳掺杂可能无法精确控制电子态和带隙的调整,导致光催化性能不均匀和不可预测。纯碳掺杂的不规则结构和亚优化电子态可能导致光催化反应的能量障碍升高。因此,迫切需要开发有效的修饰方法以克服碳掺杂g-C3N4的局限性。通过将氮替换为碳的路径实现碳掺杂,能够得到更均匀和对称的结构,减少缺陷和不规则性,从而避免纯碳掺杂g-C3N4的缺点。此外,由于碳的价电子比氮多,氮的对称替换为碳在理论上会导致电子密度的重新分布,集中π电子云,形成富π电子域。这些域展现出高电子迁移率,有效降低了窄带隙内的库仑力,并促进了激子分离。此外,富π电子域也可以作为活性位点,有效吸附和激活氧分子(O2),促进O2分子向反应性氧物种的转化,从而促进H2O2的生成。
然而,在光催化过程中引入富π电子域可能加速某些副反应。在特定反应条件下,这些副反应的速率可能超过目标反应的速率,导致副产物的积累。此外,富π电子域可能导致材料表面反应位点与反应物之间的竞争,阻碍某些反应物的有效吸附和反应,从而影响整体反应效率。值得注意的是,表面修饰技术,如引入适当的功能团(如羟基),能够显著增强反应物在材料表面的选择性吸附,从而有效减轻副反应。
基于此,江苏科技大学郭峰/施伟龙副教授课题组提出了一种巧妙的催化剂设计策略,通过尿素和酒石酸混合溶液的热解,解决由于前驱体热解过程中平台温度变化引起的多相混合材料形成问题。该方法同时在g-C3N4结构中引入富含π电子的域和亲水羟基,最终形成酒石酸改性g-C3N4(TA-CN)。富π电子域有效克服了由于增强的库仑力导致的低激子解离限制,同时提高了光生电子的迁移速率,使其能够更快地从产生位点转移到反应位点,从而增强光催化效率。此外,羟基通过其亲水性调节反应动力学,增加反应物的选择性吸附并稳定中间体,有利于目标反应。同时,羟基还促进了电荷载流子从三嗪框架向表面含氧基团的迁移,促进电荷分离,促进电子流动。这种g-C3N4框架的双重修饰策略不仅显著减少了副产物的形成,还显著提高了目标反应的选择性和效率,为光催化H2O2生产等应用提供了高效稳定的催化剂设计。
该工作以“Incorporation of hydroxyl groups and π-rich electron domains into g-C3N4 framework for boosted sacrificial agent-free photocatalytic H2O2 production”为题发表在《Chemical Engineering Journal》。江苏科技大学硕士研究生沈钰为本文第一作者,江苏科技大学郭峰和施伟龙副教授为本文通讯作者。
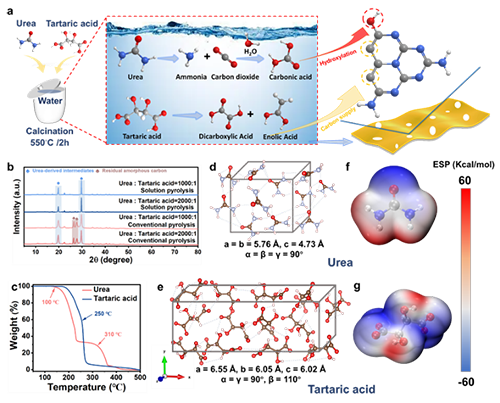
图1 (a) 溶液热解法制备TA-CN样品的示意图。(b) 尿素和酒石酸混合物在300 ℃下进行常规和溶液热解20 min后的XRD模式。(c) 空气条件下尿素和酒石酸的TGA曲线。 (d) 尿素和 (e) 酒石酸的晶体细胞结构。(f) 尿素和 (g) 酒石酸的Mulliken电荷分析。
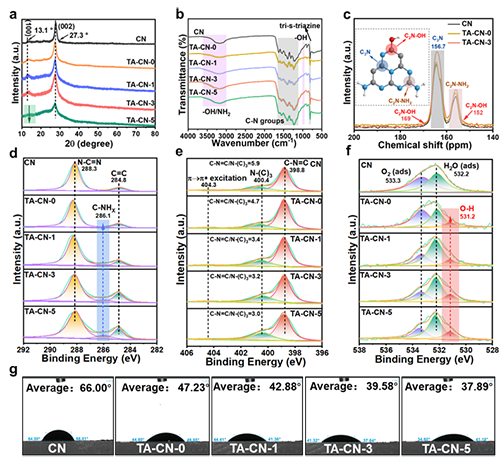
图2 (a)所有样品的XRD图谱;(b)所有样品的FT-IR光谱;(c)CN、TA-CN-0和TA-CN-3的NMR光谱;(d)C 1s的高分辨率XPS光谱;(e)N 1s的高分辨率XPS光谱;(f)O 1s的高分辨率XPS光谱;(g)所有样品的水接触角。

图3(a)CN的TEM图像;(b)TA-CN-0的TEM图像;(c)TA-CN-1的TEM图像;(d)TA-CN-3的TEM图像;(e)TA-CN-5的TEM图像;(f)TA-CN-3的高角度环形暗场(HAADF)及元素映射图像。

图4 (a)UV–vis-NIR吸收光谱;(b)带隙值;(c)VB-XPS光谱;(d)所制备样品的能带结构;(e, h)2D伪彩色表示;(f, i)在350 nm激发下,CN和TA-CN-3的瞬态吸收光谱观察到的瞬态漫反射光谱;(g)CN和TA-CN-3的飞秒分辨瞬态吸收动力学及相应的拟合结果;(j)CN和TA-CN-3的时间分辨光致发光(TRPL)光谱。
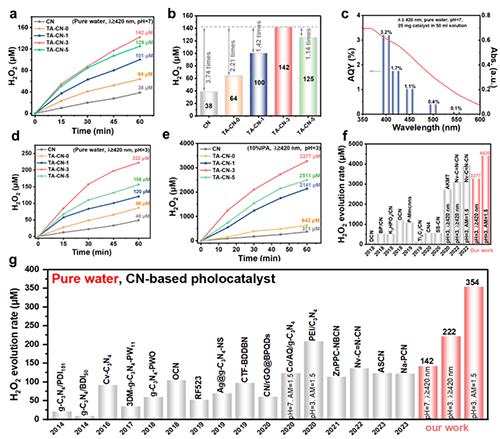
图5 (a)所合成光催化剂在pH=7的纯水中可见光照射下的H2O2产量;(b)H2O2生成速率;(c)TA-CN-3在纯水中光催化H2O2生成的量子产率(AQY)测试;(d)在pH=3的纯水中,无添加牺牲剂的H2O2产量;(e)添加牺牲剂的H2O2产量;(f)TA-CN-3与其他基于g-C3N4的光催化剂在含有牺牲剂的系统中的H2O2生成性能比较;(g)在纯水系统中的H2O2生成性能比较。

图6 (a)在1600 rpm下,CN和TA-CN-3在O2饱和的0.1 M KOH中测得的RRDE极化曲线,环电流(上部分)和盘电流(下部分);(b)计算的平均转移电子数;(c)应用电压下的H2O2选择性;(d)CN和TA-CN-3的温度程序化O2脱附(TPD-O2)曲线;(e)在DMPO存在下,CN和TA-CN-3的O2 EPR信号;(f)TA-CN-3在纯水中光催化H2O2生成过程中的原位FT-IR光谱。

图7 (a, e)结构模型;(b, f)部分态密度(PDOS);(c, g)CN和TA-CN的HOMO电荷密度分布;(d, h)CN和TA-CN的LUMO电荷密度分布;(i)CN和TA-CN上光催化H2O2生成反应的自由能谱。

图8 TA-CN光催化剂中羟基和富含π电子域协同促进光催化H2O2生成的示意图。
原文链接:https://doi.org/10.1016/j.cej.2024.155774
- 中科院化学所张金明、北京大学赵清团队 Adv. Mater.:结晶路径优化与高指数晶面稳定实现高效稳定的钙钛矿光伏 2026-06-19
- 南京大学徐凝/朱嘉 AFM:用于高效稳定蒸发冷却的氟弹性体界面改性吸湿水凝胶 2026-03-20
- 华南理工王志明、港科大林荣业/唐本忠 AM:涉及热激子的双通道阶梯能量转移策略构建具有窄发射和高亮度特性的高效稳定蓝色OLED 2025-03-29
- 中国科大陈昶乐、邹陈/安大王福周 Nat. Commun.:催化剂赋能抗氧策略实现本征抗氧化聚烯烃的便捷合成 2026-07-11
- 长春应化所陶友华研究员团队 Angew:共价硼烷-硫脲催化剂实现交硫酯立构保持开环聚合 2026-07-07
- 中科大陈昶乐、邹陈/河北工大刘宾元 JACS:离子簇催化剂组装策略构建烯烃聚合通用调控平台 2026-07-02
