将温室气体二氧化碳转化为高附加值材料,是实现碳中和目标的重要路径之一。近日,杭州师范大学材化学院李博副教授与浙江大学高分子系伍广朋团队合作,通过精准设计咪唑鎓功能化的双功能有机硼催化剂,实现了二氧化碳与环氧化物的高效共聚合,为发展新型无金属催化体系提供了新路径。
2021年,浙江大学伍广朋课题组首次提出“分子内动态路易斯酸碱多核催化体系”(DLMCS)概念(Acc. Chem. Res. 2021, 54, 4434–4448)。该体系的核心设计包含四个关键要素:可调控的路易斯酸性硼中心、可调控的抗衡阴离子、作为“阴离子中转站”的季铵/鏻阳离子,以及连接硼中心与阳离子的柔性碳链。通过分子内协同作用,该设计有效克服了传统双组分催化剂在稀释条件下因熵减导致的活性下降问题,从而实现了二氧化碳催化转化的高活性与高选择性。基于这一理念,团队发展出一类兼具高活性、模块化可设计性及规模化制备潜力的有机硼催化体系。
相关研究以Copolymerization of CO2 with Epoxides by Imidazolium Engineering in Bifunctional Organoboron Catalysts为题发表在Macromolecules上。杭州师范大学硕士研究生栗薇和浙江大学博士研究生吴天昊为文章的第一作者,李博副教授和伍广朋教授为文章的通讯作者。
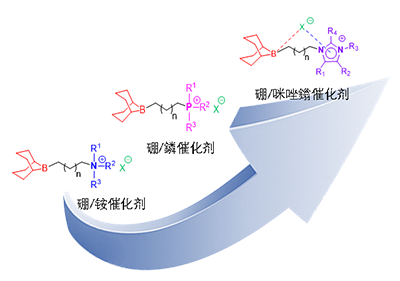
图1. 可模块化设计的有机硼/铵鎓盐、有机硼/鏻鎓盐和有机硼/咪唑鎓盐三类催化剂
尽管上述已报道的有机硼催化体系展现出优异的催化活性与可调控性,但其结构与性能之间的构效关系,尤其是对催化活性具有关键影响的阳离子部分,仍缺乏系统性研究。例如,2022年该团队通过系统对比磷系与氮系有机硼催化剂,首次证实阳离子结构的精细调控对催化性能具有决定性作用(Macromolecules, 2022, 55, 6443,图1)。然而,无论是季铵盐还是季鏻盐阳离子,其电子效应与空间结构的调控手段仍较为有限,导致阳离子的电子特性与立体效应如何协同调控催化效率,尚未得到深入系统的解析(Macromolecules 2024, 57, 8957)。
基于前期研究基础,杭州师范大学李博副教授与浙江大学伍广朋研究课题组近期提出了以咪唑鎓阳离子替代传统铵基或磷基阳离子,构建了一系列新型双功能有机硼催化剂。咪唑鎓阳离子具备独特的π共轭骨架,正电荷离域于整个杂环体系,可通过精准引入不同取代基,实现对其电子性质与空间位阻的协同调控,从而为系统揭示“阳离子结构–催化性能”关系提供了理想平台(Macromolecules, 2026, 59, 1251–1261,图1)。
研究团队通过调控咪唑环上取代基的种类与位置,首次阐明了咪唑鎓阳离子的π共轭效应对阴离子迁移行为的调控机制,并实现了活性位点间阴离子转移过程的精确调控;同时,首次发现并报道了硼中心配位效应与咪唑环电荷效应之间对活性阴离子的竞争性作用,该竞争直接影响聚合反应路径与动力学行为。此外,得益于咪唑鎓特有的π共轭效应,该类催化剂展现出区别于传统季铵盐和季鏻盐体系的独特聚合行为,兼具更宽的温度适用窗口(25–120 ℃)与更温和可控的聚合速率(TOF > 500 h-1)。本工作充分揭示了双功能有机硼催化体系中阳离子组分的高度可模块化设计性与可调性。
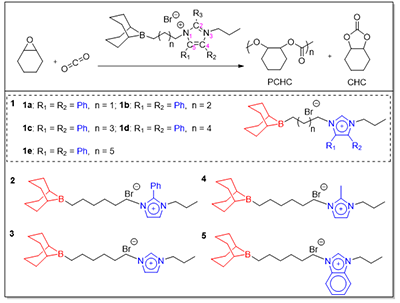
图2. 本工作研究的含咪唑有机硼催化剂的化学结构
本研究中,团队设计了两类新型催化剂(图2):系列一1a–1e:系统调控硼中心与咪唑鎓阳离子之间的柔性亚烷基链长度(C3–C7);系列二2–5:在咪唑环的C2、C4及C5位引入不同取代基(苯基、氢、甲基、苯并基)。实验结果表明:碳链长度对催化活性具有显著影响:以六亚甲基为连接单元的催化剂1d表现出最优活性;链长过短(C3–C5)或过长(C7)均导致活性下降,证实适配的空间距离是实现硼中心与咪唑鎓协同催化的关键结构因素。C2位取代基效应突出:C2位引入苯基的催化剂2活性最高,显著优于未取代的催化剂3;而甲基取代的催化剂4亦展现出明显提升的活性,表明适度的给电子性和空间位阻有利于催化性能。苯并稠合显著抑制活性:苯并咪唑鎓结构的催化剂5几乎失活,说明过度共轭削弱了咪唑鎓的路易斯酸性或阻碍了关键中间体的形成,从而干扰催化循环。
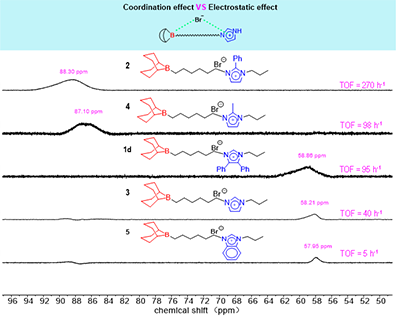
图3. 催化剂1D、2、3、4和5在CDCl3中的11B NMR(192 MHz,25℃)
11B NMR化学位移的系统性变化为理解所观察到的催化活性差异提供了直接的光谱依据(图3)。11B NMR分析表明,催化剂活性与其硼中心的化学位移密切相关:活性最高的催化剂2(δ = 88.30 ppm)呈现显著的低场位移,反映出咪唑鎓阳离子通过电子效应显著降低了硼中心的电子密度。催化性能主要取决于取代基诱导的π共轭效应及由此引发的电子重新分布。其中,N-1位孤对电子参与芳香离域,削弱了氮原子的电子密度,并在双功能催化体系中产生跨空间的电子传递效应。
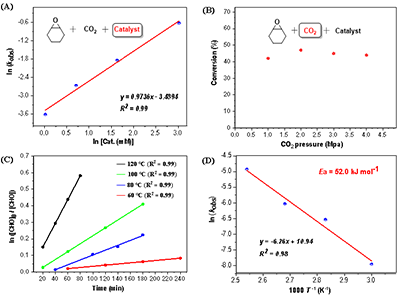
图4. 动力学测试
动力学研究表明:反应对催化剂浓度呈一级依赖;对二氧化碳压力呈零级依赖,说明二氧化碳插入非速控步;对环氧化物浓度呈一级依赖;反应表观活化能 (Ea = 52.0 kJ mol-1),与金属催化剂相当(图4)。
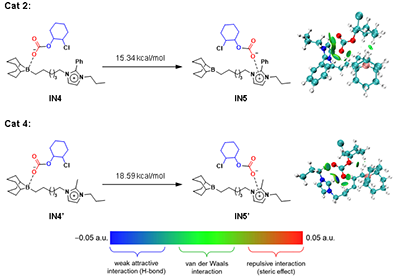
图5. 催化剂2和4关键中间体链转移的吉布斯自由能
DFT计算进一步揭示:催化剂2中苯基取代增强了π共轭,优化了碳酸根阴离子从硼中心向咪唑鎓的转移过程(ΔG = 15.34 kcal/mol),而甲基取代的催化剂4则需更高的能量(ΔG = 18.59 kcal/mol),进一步验证了实验结果(图5)。
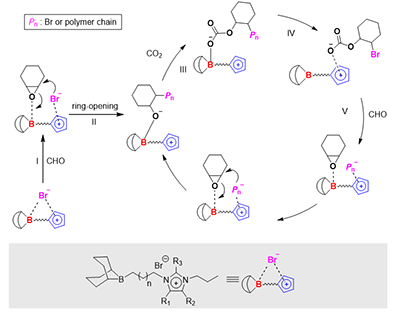
图6. 催化剂催化环氧环己烷/二氧化碳共聚的反应机理
基于实验与计算,团队提出分子内协同催化机制:环氧化物与硼中心配位(I),被Br-亲核开环(II);二氧化碳插入形成碳酸根阴离子(III);碳酸根转移至咪唑鎓阳离子(IV),空出的硼中心活化下一分子环氧化物(V);碳酸根回攻开环,完成链增长。聚合过程中咪唑鎓充当“阴离子中转站”,通过静电作用将阴离子限域在硼中心附近,从而实现高效协同催化。
本研究通过系统调控咪唑鎓阳离子的电子结构与空间位阻,阐明了双功能有机硼催化剂中阳离子组分在协同活化与立体控制中的关键作用,为无金属催化剂的理性设计提供了新范式。所开发的催化剂兼具高活性、高选择性及优异热稳定性,可在温和条件下高效催化二氧化碳与环氧单体的共聚反应,为可持续聚碳酸酯材料的绿色合成提供了新路径。未来,该催化体系有望拓展至多种功能性环氧单体,实现结构可控、性能可调的功能化聚碳酸酯的精准合成。
论文信息:Li, W.; Wu, T.; Li, B.; Wu, G.-P. Copolymerization of CO2 with Epoxides by Imidazolium Engineering in Bifunctional Organoboron Catalysts.
Macromolecules 2026, 59 (3), 1251-1261.
https://doi.org/10.1021/acs.macromol.5c03252
- 陕科大王学川教授/党旭岗副教授JBB:壳聚糖-羧甲基纤维素气凝胶对含Cr(Ⅲ)、Al(Ⅲ)和Zr(Ⅳ)制革废水的修复及资源化利用 2024-11-25
- “废塑料橡胶资源利用技术与新装备开发研讨会”成功在京举办 2009-06-18
