序:风起云涌的机理之争
1995 年,对于整个高分子化学界来说,是很重要的一年。这一年,美国Carnegie Mellon University(卡内基梅隆大学)的Krzysztof Matyjaszewski和王錦山在J. Am. Chem. Soc. 1995, 117, 5614 上发表了划时代的工作,首次发现和提出了著名的原子转移自由基聚合(ATRP,如图1)。
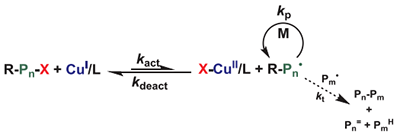
图1 原子转移自由基聚合(ATRP)的机理
ATRP 的基本原理是,引发剂(烷基卤素,如2-溴异丁酸乙酯,EBriB)与铜金属配合物CuI/L 反应,生成 CuII/L和自由基,同时将卤素原子转移到过渡金属配合物上;随后生成的自由基攻击乙烯基单体。同时,过渡金属配合物 CuIIX/L 使自由基失活并将卤素原子转移回去,形成休眠种并再生过渡金属配合物 CuI/L。在此之后数年间,ATRP 体系迎来爆发式发展:不同的催化剂、配体、引发剂等新体系如雨后春笋般涌现。
1997 年,金属铜- Cu(0) 首次被引入 ATRP 体系。Cu(0) 的出现,一方面带来了明显优势 - 聚合速度更快、催化剂用量更低等;另一方面,也把原本“只需要考虑 CuI 与 CuII”的体系,变成了 CuI / CuII / Cu0 三种价态共存的复杂系统。机理上的争论,自此埋下伏笔。
2006 年,美国University of Pennsylvania(宾夕法尼亚大学)的 Virgil Percec教授在美国化学会志(Journal of the American Chemical Society)上发文“Ultrafast Synthesis of Ultrahigh Molar Mass Polymers by Metal-Catalyzed Living Radical Polymerization of Acrylates, Methacrylates, and Vinyl Chloride Mediated by SET at 25 °C”(JACS. 2006, 43, 14156),这项工作中实现了多种单体的精确可控聚合,并正式提出了针对Cu0催化体系的单电子转移活性自由基聚合(SET-LRP)机理(图2)。SET-LRP认为在极性溶剂中,Cu0 作为引发剂的唯一活化剂,可与烷基卤素引发剂发生外层电子转移(outer-sphere electron transfer,OSET)反应,该过程生成自由基和 CuI。生成的自由基引发单体并发生链增长,而形成的CuI 会“自发”发生歧化反应(disproportionation),产生具有高度活性的“新生” Cu0(作为活化剂)以及CuII(作为钝化剂)。在这一过程中,归中反应(comproportionation)被认为几乎不起作用。该观点彼时也得到了英国University of Warwick(华威大学) David Haddleton教授等人的大力支持。
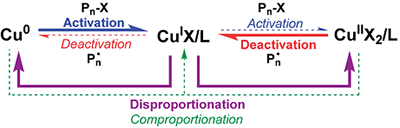
图2 单电子转移活性自由基聚合(SET-LRP)的机理
紧接着,ATRP 创始人 Matyjaszewski 教授在 2007 年在大分子(Macromolecules)上发表了“Role of Cu0 in Controlled/“Living” Radical Polymerization”( Macromolecules 2007, 40, 22, 7795),提出了补充活化剂和还原剂的原子转移自由基聚合(SARA ATRP)机理(图3),对 Cu0在体系中的作用给出另一套相反的解释。SARA ATRP认为烷基卤素首先由 CuI 进行活化,随后由 CuII 进行钝化,从而建立起可逆的活化/钝化平衡。在这一体系中,Cu0主要承担补充活化剂以及通过归中反应还原 CuII 的作用。相比之下,歧化反应在该过程中贡献极小。所有烷基卤素的活化主要由 CuI完成,并且该过程是通过内层电子转移机制(inner-sphere electron transfer,ISET)来实现的。自此,围绕Cu0催化体系的“机理之争”正式拉开帷幕。不同的理论模型,乃至一些折中或过渡性的解释相继被提出(例如 Harrisson 等认为,在特定条件下歧化与归中过程可以同时存在, Macromolecules 2012, 45, 7388)。然而,尽管观点纷呈,这些模型始终未能形成统一框架,也始终没有任何一种机理能够全面、系统地解释Cu(0)催化体系中所观察到的全部实验现象。
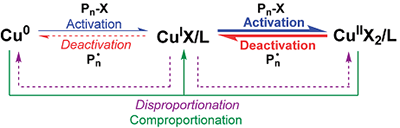
图3 补充活化剂和还原剂的原子转移自由基聚合(SARA ATRP)的机理
一次“涨不上去”的转化率,埋下长年的困惑
2012 年,王文新教授指导的博士生赵天宇(现中山大学副教授),在一次看似普通的ATRP聚合实验中(丙烯酸甲酯 (MA) /EBriB/Cu(I)/Me6TREN/25 oC/DMSO,如图4),遇到了一个“说不通”的结果:聚合反应中间会停止,转化率始终涨不上去,并且发现反应体系中发现有新生的Cu0。王教授第一反应是:想要相信实验,需要确认实验。于是,他让学生再做一遍。第二次实验,结果依旧;第三次重复,还是一样。“这不应该啊。” 面对一个在文献中被视为“gold standard”的体系,为什么在自己实验室里却出现了如此异常的行为?那段时间里,王教授心中反复权衡:是不是学生实验操作有纰漏?还是说,文献中ATRP的机理模型本身就存在某些被忽略的问题?我们看到的是“偶然例外”,还是“规律的一角”?尽管多次重复实验,但在当时仍难以百分之百排除所有实验误差的可能。于是,这个问题暂时被放下,却从此一直萦绕在王教授心中——像一颗埋在土里的种子,等待被再一次唤醒。
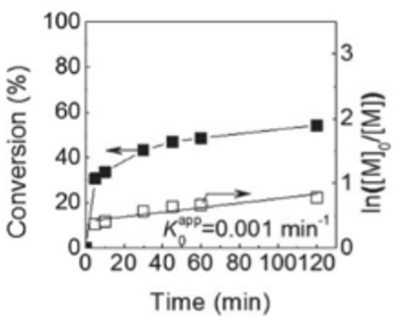
图4 ATRP体系下丙烯酸甲酯 (MA) 聚合转化率在约50% 后难以继续增长,条件-MA/EBriB/Cu(I)/Me6TREN/25 oC/DMSO
第二位博士生的“同样异常”,让怀疑更加坚定
2014 年,另一位博士生高永胜(现University of Texas at Dallas助理教授)在进行相关实验时,也观察到了极为相似的现象:转化率又一次“卡”在那儿,上不去。当第二份互相印证的数据摆在眼前时,2012 年的那份不安与困惑再次涌上心头。王教授这一次不再满足于“也许是偶然”这样的解释。他和高永胜决定“打破砂锅问到底”:多次重复、对照条件、排查操作、检查仪器…… 结果却非常一致——曲线一遍遍地重复着同样的形状,“涨不上去”的聚合反应行为一再出现。这一次,他们确信:实验是对的,问题应该是出在机理上。在这看似最为经典机理模型之下,实际上可能存在着巨大的漏洞。经过细致的分析和思考,再回头看 2006–2007 年的机理之争,其争论的核心问题也变得清晰起来——可以总结为以下三点:
-
一价铜,二价铜,零价铜在反应过程中的作用分别是什么?
-
不同铜种之间是如何相互转化的?
-
是否存在一新的通用机理?
要解答这些问题,一个行之有效的切入点,是从反应体系中至关重要的配体体系入手。已有大量文献报道,两种氮配位多齿配体均已成功应用于ATRP 体系中:一种是PMDETA(N,N,N’,N’,N’’-pentamethyldiethylenetriamine),另一种是Me6TREN(tris[2-(dimethylamino)ethyl]amine)。在这些体系中,均可实现如 PMA 等聚合物的可控聚合。然而,当时,在 SET-LRP 体系中,以 PMDETA 作为配体对 MA进行聚合的研究报道极为有限,绝大多数相关工作均采用 Me6TREN作为配体。因此,王教授和高永胜想,如果将 PMDETA 这一在 ATRP 反应体系中被广泛使用的配体引入 SET-LRP 体系用于 MA 的聚合,其结果有望为反应背后的机理提供新的认识,最终有望进一步检验该体系/乃至更多体系究竟遵循 SARA ATRP 机理还是 SET-LRP 机理。基于上述假设,他们设计了一系列实验,采用 PMDETA 作为配体,并分别与不同价态的铜物种组合形成催化体系,用于 MA的聚合反应。结果发现Cu0/PMDETA(MA/EBriB/DMSO/25 oC)体系的聚合控制性明显变差,早期呈现出类似FRP的不可控增长。但若在反应开始加入少量CuII,即可获得可控聚合。仅仅是更换配体,就引发了机理行为的巨大差异,这又一次提醒他们:传统的ATRP和SET-LRP机理模型远远不够解释这些实验现象和结果。
很明显,在这个Cu0/PMDETA 体系,在反应早期,这种类自由基聚合(FRP)的不可控聚合,肯定是Cu0 的瞬间活化导致。这符合SET-LRP(Cu0可以迅速激活休眠物种),但是和SARA ATRP不符(Cu0的活化过程应该是非常缓慢)。另一方面,反应中后期:钝化效果逐渐出现,这必然是CuII的逐渐积累导致,这符合SARA ATRP(CuII 是由 CuI 活化形成的),但是和SET-LRP不符(CuII 是由 CuI 瞬间发生歧化反应生成的)。王文新教授让高永胜同时也尝试了添加CuII(Cu?&CuII/PMDETA) 体系,他们发现添加CuII后反应初期即表现出良好的可控性,聚合控制得到很大改善。因此,该体系的聚合行为也符合SARA ATRP,但是和SET-LRP不符。所以,可以看出,无论是哪一种反应机理,都只能解释一部分现象,却没法理解不同反应条件下的整个聚合过程。
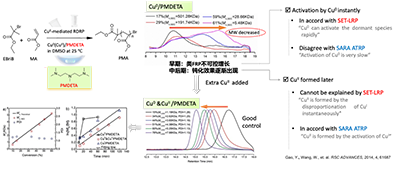
图5 Cu0(&CuII)/ PMDETA 催化MA体系的聚合现象与 SEL-LRP 和 SARA ATRP 模型的相同点和相悖点
经过细致的分析和思考,王教授和高永胜认为:在 Cu(0)&CuII/ PMDETA 催化体系中所得到的可控聚合,其真实机理偏离了传统 ATRP 和 SET-LRP 的典型描述,更合理的解释应当是两种经典机理之间的竞争与平衡所共同塑造的结果。
他们怀着极大的兴奋,将这一思考整理成题为“Are the SARA ATRP and SET-LRP coexisted?”的文章,期待能与领域同行分享这一新视角。然而,现实给他们泼了一盆又一盆冷水。
多次被拒与尖锐评语:真正的“艰辛”,从这里开始
挑战经典或传统框架的工作一开始总是难以被大家所接受的,这篇工作在投稿过程中屡遭拒稿,审稿意见更是异常严厉:“It is still the SARA ATRP…” “It is still the SET-LRP…” “The authors have no understanding of ATRP or SET-LRP…” 甚至还有:“This is a terrible paper that should never be published anywhere…”,这样的评价对大家无疑是沉重的打击。王教授和团队几经推演、谨慎论证,结果却被一句“terrible paper”否定得一文不值。从满心欢喜到彻底冰凉,甚至伤心落泪(高永胜作为初入科研领域的博士生,面对此挫折深感挫败),这是每一个科研工作者都可能经历的一课。
但这些评价反而进一步激发了王教授及团队对真实机理的探索。王教授并没有因为几次尖刻的评语就放弃。他心里非常清楚:当实验现象一次次重复出现时,科学家最应该坚持的是对“真相”的追问,而不是对权威的盲从。他一边安慰学生,一边继续思考:如果我们的数据没错,那说明现有机理的确解释不全;如果解释不全,那总有一天要有人把这件事说清楚。
在这里,我们也讲一个轶事,2015年,王文新教授邀请Karen Wooley教授(当时Karen是JACS杂志的副主编)作为外审参加王教授的博士生赵天宇的博士答辩。在此期间,王文新教授和Wooley教授提到了他们最近在Cu(0)活性自由基聚合催化体系的研究工作。Wooley教授很惊讶且直率地和王教授开玩笑地说:“Wenxin, you are really stupid to enter into this muddy field. You should know there has been too much heated argument here, it is not worth it and even bad for you.”。
把目光投向“诱导期”:从局部现象到统一机理
随着研究的深入,王教授他们同时也注意到Cu(0)活性自由基聚合体系中一个很有意思的现象——诱导期(induction period) - 单体转化率几乎为零。
自 2010 年起, Haddleton 等人报道,在经典Cu(0)/EBriB/Me6TREN/ DMSO/25 °C聚合MA的体系中,聚合开始前存在约 30 min 诱导期(Polym. Chem., 2010, 1, 1086)。一时间,关于诱导期成因的解释层出不穷:
-
Haddleton 认为:诱导期长短主要由铜线表面积决定,面积越大,诱导期越短(Polym. Chem., 2010, 1, 1086);
-
Percec 则认为:诱导期源于铜线表面的氧化铜层(Cu2O),去除该氧化层即可“消除诱导期”(J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 5109)。
然而在实际实验中,王教授和高永胜发现:即便通过完善的预处理去除了 Cu2O,诱导期依然存在,反而,加入二价铜就完全可以消除诱导期,这种实验现象都无法被以上两种观点所解释(图6)。
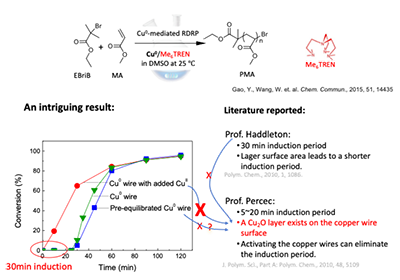
图6 诱导期及其相关解释
这无疑说明:诱导期绝不只是一个简单的“表面氧化层”问题。于是,他们设计了一系列不同铜种催化剂体系的实验,进行系统的分析和推理(图7),并最终得出结论:诱导期和随后的自加速聚合反应源于不同铜物种(CuI 和 CuII)的相对积累。此初步研究成果发表在Chem. Commun., 2015, 51, 14435.
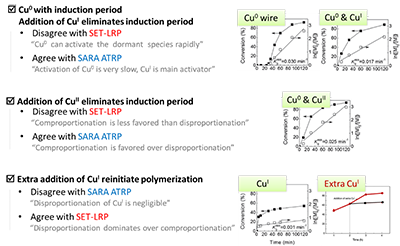
图7 不同体系下的聚合现象与 SEL-LRP 和 SARA ATRP 的相同点和相悖点
王教授随后将这一重要进展通过邮件分享给 ATRP 的创始人 Matyjaszewski 教授,然而,Matyjaszewski 教授回信道:“Simulations and experiments of SARA ATRP under similar conditions but using EBrP(2-溴丙酸乙酯)/MBrP(2-溴丙酸甲酯)(instead of EBriB) as initiator do not show any induction period.” 这条信息,再次让整个问题笼罩上一层新的迷雾:诱导期真的被解释清楚了吗?引发剂活性、不同价态铜种之间的转化到底存在怎样更深层次的联系?针对这一现象,王教授认为那就继续通过实验来探索EBrP/MBrP 的体系。
从“现象解释”走向“统一模型”
虽然历经质疑与挫折,但王教授没有放弃,他持续关注并思考着铜活性自由基聚合体系的机理本质。经过多年积累与沉淀,王教授与博士生吕京(现University College Dublin都柏林大学助理教授)和硕士研究生苗永鹏设计并系统研究了多种组合。不同催化体系:CuI、Cu(0)、Cu(0)&CuII,不同引发剂体系:α-溴异丁酸乙酯(EBriB),EBrP, MBrP,2-氯丙酸甲酯(MClP)。随后通过严谨的分析,他们对“诱导期出现与否”的本质有了清晰的认识,并归纳出了一套引发剂活性对诱导期影响的规律。总的来说,在 Cu(0)介导的 MA 可控活性聚合中,诱导期出现与否,源于反应早期的歧化/归中程度(导致 CuI 和CuII累积量的差异)。而不同活性的引发剂,自然会直接影响不同铜种浓度在早期的积累。由此得出,真实的机理并不是SET-LRP 或 SARA ATRP单独可以解释的,应该是两者综合作用的结果。在铜活性自由基聚合体系中,歧化与归中同时存在,应该同时被考虑(图8)。
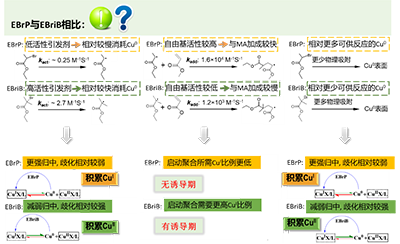
图8 引发剂的活性如何影响聚合诱导期
这项工作先后投递至JACS, Macromolecules, Chemical Communications以及Polymer Chemistry,但均被拒稿,审稿意见言辞犀利:“…How is this different from SARA ATRP…” “…The conclusions drawn are already well known…” “…I don’t see this manuscript adding anything meaningful to the conversation…” “…The experiments do seem to be technically sound. However, without more direct kinetic characterization of the copper oxidation states…” 面对这些质疑,王教授团队并未止步,而是逐条回应审稿人的意见,反复补充对照实验、深化机理分析并多次修订稿件。然而,虽然王教授等提供了针对审稿人和编辑邮件的所有实验证据,多数编辑和审稿人还是以“这个机理的工作已经不是研究热点或者不存在问题”为由强行拒稿。最终,王教授等人只能先将这项工作发表在了Polymer, 280, 2023, 126055,毕竟期刊不重要,重要的是让真正的Science为大家所知。至此,错综复杂的铜活性自由基聚合体系机理被第一次阐明清楚。王教授团队认为,无论ATRP 还是 SET-LRP,其实都应该在新的机理框架下加以统一理解,并将这一体系概括为——铜基可逆钝化自由基聚合(Cu-based RDRP)。其中的核心在于:不同铜物种之间的转化过程(歧化/归中)与聚合平衡需要被同时考虑。这一工作发表后,领域内不少资深专家主动与王教授交流。Haddleton 课题组的 Dr. Levere 在交流中提到,“我非常认真地读了这篇关于聚合诱导期的文章,这项工作让我们早年得到的实验结果“重新焕发了意义”。
从最初的审稿“重锤”反对,到如今来自一线同行的由衷认可,这其中的落差,只有真正经历过的人才懂。
从机理走向应用:甲基丙烯酸甲酯(MMA)、苯乙烯(St)、丙烯酰胺(AM)的精准可控聚合被逐一攻克
机理框架逐渐清晰后,下一个自然的问题就是:能否利用深刻理解建立的新机理,反过来指导精准控制聚合体系设计,解决具体的精准可控聚合难题?
1. 低活性单体——MMA,St
MMA 作为典型的低活性单体,其可控聚合长期存在拖尾等问题。王教授课题组根据前述机理解读出一条关键思路:低活性单体的体系中,聚合平衡会相对更慢, 铜种之间相互转化时间相对更长,所以CuII 主要消耗于铜种之间转化平衡 - 归中过程,而非作为钝化剂参与聚合平衡。因此,单纯外加少量 CuII,并不能显著改善聚合可控性。
反过来看,引发剂的选择就变得尤为关键。基于Polymer, 280, 2023, 126055的结论,为了在聚合早期增强钝化效果,应该使用更高活性的引发剂。一方面,CuII在早期得到积累。另一方面,更高的脱溴能力以及引发剂自由基稳定性,可以降低实现可控对CuII比例的要求。于是,王教授组选择了更高活性的引发剂甲基α-溴苯乙酸酯(MBPA),在仅加入 5% CuII的条件下,就实现了良好的可控聚合。
对于 St,传统条件下在低活性引发剂(PEBr/MBrP)和高活性配体(Me6TREN)组合下,仍然存在明显的拖尾问题。王教授团队依照在 MMA 上总结的思路:1)使用高活性的 MBPA 提升早期 CuII的有效积累;2)使用钝化效果更强的溶剂甲苯,进一步强化钝化作用。最终,也成功实现了 St 的精准可控聚合,该工作发表于Chin. J. Polym. Sci. 2019, 37, 591。
2. 高活性单体——AM
在解决了 MMA 和 St 之后,王教授与博士生李子山(现曼彻斯特大学博士后)和吕京博士把目光投向了更具挑战性的高活性单体 AM。一方面,聚丙烯酰胺是一类极其重要的功能高分子材料,在外科填充、采油以及土壤调理等领域具有广泛应用;另一方面,铜活性自由基聚合体系长期未能实现 AM 的可控聚合。因此,铜催化条件下 AM 的可控聚合一直以来备受关注。然而,基于铜催化的 AM 聚合长期以来存在以下问题:转化率偏低、分子量分布偏宽等。根本原因在于:丙烯酰胺类单体活性极高,副反应众多;且单体上的酰胺基团能够与铜种配位,降低铜催化活性。
基于此前建立的机理框架,王教授团队清楚地意识到引发剂的选择在这里尤为关键。于是他们选择了与 AM 结构相似的2-溴丙酰胺(BPA)作为引发剂,以提供高效的快引发。另外,其水溶性好、价格低廉。同时,他们发现 AM 体系聚合极快、终止与副反应严重,少量CuII的加入无法提供有效的控制,并保持持续的链增长。通过一系列实验的摸索,他们终于发现:需要大量钝化剂,也就是说,在大量钝化剂(40% )CuII才可以实现低 PDI,高转化率的可控聚合。在此条件下,团队不仅成功实现了 AM 的可控聚合,也实现了 PAM嵌段聚合物的可控聚合,验证了聚合物链末端的活性。此工作最终发表于Macromolecules 2023, 56, 5111。
随后,团队继续攻克另外两种丙烯酰胺类单体:N,N-二甲基丙烯酰胺(DMA,其铜活性自由基聚合体系一直没有被成功实现)和N-异丙基丙烯酰胺(NIPAM,其Cu(0)催化的活性自由基聚合体系一直没有被成功实现)。对于NIPAM,为了在使用相同反应组分的情况下实现可控聚合,需要提供更强的钝化能力,即采用 Cu(0) 与 80% CuBr? 的组合。此外,在目标为高分子量PNIPAM 的合成时,必须采用较低的反应温度(4 oC),以避免因低临界溶解温度(LCST) 相转变而导致的聚合控制的受损。对于 DMA,与 AM 和 NIPAM 不同,实现可控聚合所需的充分钝化并不能仅通过简单增加钝化剂(即 CuII )的用量来实现;相反,获得增强的钝化效果,必须在综合考虑并合理调控其他反应参数的前提下方能实现,例如将 CuBr? 更换为 CuCl?。此外,为了在反应初期积累更多CuI以确保持续的链增长,还需要使用活性较低的引发剂2-氯丙酸甲酯 (Methyl 2-chloropropionate, MCP)。以上工作最终分别发表于Chin. J. Polym. Sci. 2023, 42, 1-6;J. Polym. Sci. 2023, 62, 1193-1200。

图9 王教授(中)正在与博士生李子山(右一)和吕京(左一)共同开展实验并进行讨论
总结与展望:从一间实验室,走向一个机理体系
回顾这一路的探索,从最初一次“转化率涨不上去”的异常开始,到多次被拒稿、饱受严厉批评,再到对诱导期的长期追踪、对 CuI / CuII / Cu0 相互转化平衡的系统梳理,以及后续对 MMA、St、AM 等系列单体可控聚合的成功应用,王文新教授团队在Cu-catalysed RDRP上的工作,逐渐形成了一套具有普适意义的机理框架,并将其命名为铜基可逆钝化自由基聚合(Cu-based RDRP):
-
可溶性铜种( CuI, CuII )浓度在聚合早期得到积累
-
配体和引发剂会改变不同铜种相互转化的动力学和其相对浓度积累
-
[CuI]/[CuII]比例是启动聚合的关键因素。
-
[CuII]/[CuI]比例是实现良好控制的聚合的关键因素。
-
歧化作用和归中作用在Cu-based RDRP中应同时考虑。
-
两种平衡共存:1) 铜种之间的相互转化平衡(歧化/归中)和 2) 聚合平衡(活化/失活,增长和终止)。
-
不同的聚合参数(如引发剂,配体,溶剂等)通过协同影响这两种平衡来影响整个聚合过程。
目前,基于这一机理的更系统性的总结性文章也在筹备之中,相信不久的将来会与大家正式见面。
回头来看,这是一条充满质疑、否定、坚持与重构的道路:从“这不对吧?”的直觉,到数据一遍遍重复后的笃定;从“terrible paper” 的冷酷评语,到同行由衷的认可与深入交流;从一个实验室里几位学生和一台 GPC、几根铜线的探索,走向对整个Cu-catalysed RDRP领域聚合反应机理的重塑与统一。
科研之路漫长而曲折,但只要始终坚持对真相的追问、对数据的尊重、对科学机理的执着打磨,终有一日,当我们回望来路,便会看到自己取得的一个个里程碑。所有的委屈、挫折与孤独,都会化作“轻舟已过万重山”的从容与愉悦。愿每一位科研工作者都能在追光的道路上,坚定前行,终见繁星满途。
王文新教授简介

王文新,教授,海外引进国家级人才,爱尔兰科学基金会(Research Ireland-RI)首席科学家,英国皇家化学会会士。安徽理工大学讲席教授,精准医学创新研究院院长,爱尔兰都柏林大学医学院查尔斯皮肤科学研究中心终身教授,并兼任都柏林大学机械和材料工程学院教授, 浙江大学求是教授。
联系方式:wwxph@aust.edu.cn
主要研究方向:
主要从事高分子可控聚合方法的前沿机理和临床应用研究,具体为高分子可控聚合机理和动力学控制策略及其在生物功能材料设计与合成中的两大方向应用:组织工程学—水凝胶以及其他功能性生物材料单独或结合干细胞疗法在骨关节炎、关节软骨缺损、骨缺损、牙科骨/软组织修复、皮肤创伤修复等医疗方向的广泛应用;基因治疗—十余年深耕研发报道多种高效聚合物结构用于基因递送,并通过可控聚合首次攻克和发明了超支化聚β氨基酯,该大类材料家族用于遗传物质的体外和体内递送,并成功应用于包括EB基因替代和编辑疗法等在内多种拥有完全自主知识产权的核酸药物的临床转化验证。
科研工作:
已发表270余篇学术论文,如Science Advances《科学进展》,Nature Communications《自然:通讯》,Nature Reviews Chemistry《自然综述:化学》,JACS《美国化学协会会刊》,Angewandte Chemie《德国应用化学》,Advanced Materials《先进材料》,Nano Letters《纳米快报》,ACS Nano《美国化学学会纳米杂志》等,并参与编纂5本学术专著的部分章节。相继被多家国际知名媒体采访和报道61次,如英国皇家学会的化学世界(RSC Chemistry World)“Polymer tied in Celtic knots”,美国的化学工艺(Chemical Processing)“Polymers Branch Out”,和美国科学日报(Science Daily)“Polymer Breakthrough Inspired by Trees and Ancient Celtic Knots”。于2010年在欧洲组织工程和再生医疗国际协会(TERMIS-EU)年会上荣获爱尔兰国家自然科学基金委颁发的再生医学领域青年科学家奖,于2011年荣获由爱尔兰国家自然科学基金委颁布的爱尔兰科学基金会首席科学家奖,2014年荣获由DEBRA协会颁发的杰出EB患者服务奖, 2025年获都柏林大学颁发的NovaUCD创新奖。先后获得了来自爱尔兰科学基金会(SFI)、欧盟(EU-FP7 & EU Horizon 2020)和其他基金单位多达1558万欧元的科研资助。被聘选为29个研究委员会和基金会的评审专家和委员会成员,其中包括中国自然基金委员会、比利时科学研究基金会佛兰德斯委员会、欧盟FP7框架和Horizon2020的玛丽居里基金会、加拿大渥太华研究基金会、葡萄牙科学基金会、捷克科学基金会、英国医疗研究委员会、荷兰科学研究委员会、以色列科学基金会、奥地利科学基金会等。被11家国际同行评议杂志聘为编委,如《纳米生物技术杂志》、《高分子》、《生物医学材料》、《材料》和《林产化学与工业》等。先后126次被国际会议和大学邀请作为主要报告人,并作为顾问、组织者、会议主席或召集人,主持了34次国际学术会议。
- 暂无相关新闻
