高熵合金由于其出色的机械性能和多样化的微观结构而受到了广泛关注。然而,由于高熵合金设计空间的维度巨大,传统的材料设计方法变得低效且昂贵,从而导致高熵合金的巨大潜力尚未充分实现。因此,理性设计策略对于在这一广阔的设计空间内有效发现具有期望性能的高熵合金至关重要。
材料的原子间势函数是对材料进行原子模拟的基础。原子间势函数的准确性也决定的原子模拟的精确度。目前高熵合金理性设计发展缓慢的原因之一就是众多原子间势函数的缺失。为了解决这一挑战,美国马里兰大学李腾教授课题组提出了一种基于图神经网络机器学习通用策略的原子间势函数的发展方案,旨在建立高熵合金中原子位置和原子力之间的复杂非线性关系,并以CrFeCoNiPd作为模型材料对这一发展方案进行了成功的展示。该方案获得的机器学习模型可以在原子力方面实现显著的预测精度(R2>0.92),且能够准确模拟高熵合金材料的力学性能和变形机理。相关工作以“A machine learning interatomic potential for high entropy alloys” 为题,近期发表于国际力学领域旗舰期刊《Journal of the Mechanics and Physics of Solids》。
图1 为基于图神经网络机器学习通用策略机器学习的原子间势函数开发过程。通过头分子动力学计算,获得合金在不同温度下原子的位置,能量,原子间的力和原子间的应力。提取原子位置信息,将位置信息和原子电负性作为机器模型的输入,原子能量为输出,对模型训练。最后根据原子的能量,计算原子间的力获得模型的力场信息。图2对比了模型预测的原子能量以及原子间的力与头分子动力学计算的原子能量以及原子间力,证实了模型具有很高的预测精度。
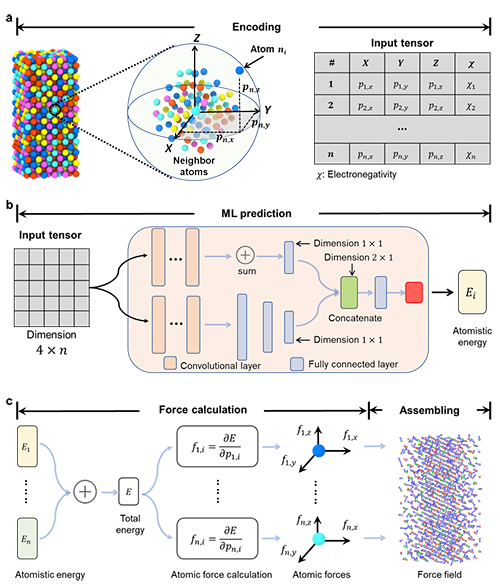
图1: 基于图神经网络机器学习通用策略机器学习的原子间势函数开发过程的示意图。a, 生成训练数据集;b, 构建机器学习模型;c, 计算原子力并组装原子力场.
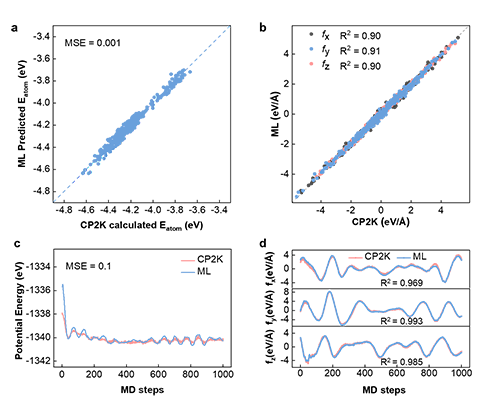
图2: 机器学习模型评估:a-b 机器学习模型预测的原子能量和原子间力与头分子动力学模拟计算结果对比;c-d 系统中第一个原子的原子力在300K下1000个动态演化过程的变化,与头分子动力学模拟计算的结果比较。
为进一步验证机器学习模型的准确性,李腾教授课题组将机器学习原子间势函数嵌入LAMMPS软件,然后进行蒙特卡罗分子动力学模拟,以获得CrFeCoNiPd 合金的稳定结构。由图3可以看出,在形成稳定结构的过程中,Pd原子有更明显的聚合现象。最后,利用机器学习原子间势函数预测的晶格常数及层错能与实验作对比如图3d和图3e所示,预测结果与实验相近。这进一步证实了机器学习势函数的准确性。
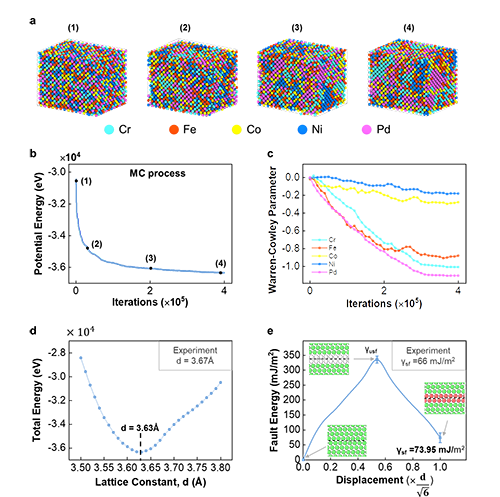
图3: 机器学习原子间势函数分析CrFeCoNiPd 高熵合金的原子结构 :a, MC模拟过程中原子结构; b, MC过程中能量变化; c, MC过程中WCP的变化; d, CrFeCoNiPd 高熵合金的晶格参数; e, 层错错能曲线。
为了证明本文发展的机器学习势函数的应用性,李腾教授课题组利用机器学习原子间势函数模拟了高熵合金 CrFeCoNiPd的压缩过程,并首次通过系统的模拟揭示了压缩过程中的变形机理其中包括位错的形成,位错的滑移及层错的生成过程。压缩模拟过程中发现的位错交滑移现象与实验观察一致。这进一步证实了机器学习势函数的实用性和准确性。模拟也揭示了高堆垛层错能在改善CrFeCoNiPd 高熵合金强度中的关键作用。
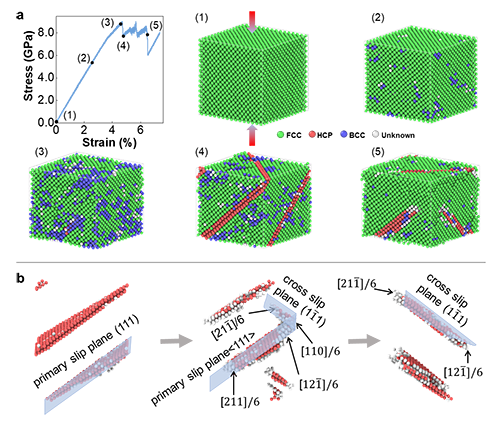
图4 基于机器学习原子间势函数的CrFeCoNiPd 合金压缩模拟。 a, 压缩应力应变曲线及不同应变下的原子图;b, 变形中的交滑移过程。
建立基于图神经网络机器学习通用策略的原子间势函数的策略为推动高熵合金的路线设计带来了新的思路,未来可能推动更高效和精确地探索更多新的高性能高熵合金。
论文链接:L Wu, T Li, A machine learning interatomic potential for high entropy alloys, Journal of the Mechanics and Physics of Solids, 105639 (2024)
https://doi.org/10.1016/j.jmps.2024.105639
- 哈工大(深圳)朱时裴教授课题组诚招硕士、博士、博士后 - 材料、化学、物理、机械、生物工程 2025-09-17
- 清华大学李晓雁副教授课题组等制备了一种克服强度-可恢复性制约的三维高熵合金—聚合物复合纳米点阵超材料 2018-07-11
- 吉林大学邹勃教授团队 Nat. Commun.:压致变色凝胶用于机器学习辅助的物理不可克隆光学防伪 2026-05-13
- 江苏大学聂仪晶教授团队 Macromolecules:分子模拟+机器学习双轮驱动 - 高强度高愈合效率氢键自愈合聚合物理性设计新范式 2026-05-06
- 港科大杨晶磊/加州理工 William A. Goddard III《ACS Catal.》:结合机器学习势函数与量子化学 - 探究界面聚合中的单分子水催化效应 2026-03-20
