摘要:双螺杆脱硫作为一种废旧橡胶脱硫的新方法、新技术,正引起越来越多的学者的关注。该脱硫方法属于一种热力双重作用脱硫方法,脱硫效果与温度和剪切力的选择密不可分。温度和剪切力提供了含硫键断裂所需的外部能量,所以含硫键断裂前后的系统能量变化问题是一个重要课题,而模型的建构则是最为关键的第一步。为此,构建了分别含2条链的天然橡胶(NR)、丁苯橡胶(SBR)以及8条链NR、SBR周期模型,其中主链与主链之间分别通过单硫、双硫、三硫键相连接(交联)。采用COMPASS力场,利用分子动力学方法,在一定的模拟条件下,对交联键断裂及未断裂系统进行了分子动力学模拟。根据分子动力学得出的能量数据,计算出交联键断裂前后的能量差。结果表明,2条链的NR模型获得的能量差数据存在较好的定性关系,在增加模拟时间和对模型通过周期性排列改善后,能量差数据更为合理。而对于2条链SBR模型和8条链SBR周期模型,由于非键能作用影响超过了断裂的键能,断裂能量数据不太稳定(出现负值且二硫键相关能量数据波动较大),说明这两种模型不太适合本课题研究。相对而言,8条链NR周期模型中平均单个键的能量差数据波动幅度不大,断裂能量差偏差较小,结果非常理想;而且,能量差大小与键能理论值顺序完全一致,说明8条链NR周期模型非常适合于研究硫化橡胶含硫键的断裂能量差。
关键词:分子动力学;硫化橡胶;交联键;断裂能
中图分类号:O641.3 文献标识码:A
文章编号:2095-0411(2012)01-0001-06
我国是世界上第一大橡胶资源消耗国,同时又是橡胶资源十分匮乏的国家,近80%的天然橡胶NR和40%以上的丁苯橡胶SBR依赖进口,其中NR的进口依存度已经高于石油、铁矿和粮食等,位列第1位。橡胶消耗量中,轮胎橡胶占总量的70%以上。据世界环境卫生组织统计,目前世界废旧轮胎积存量已达30多亿条,并以每年约15亿条的数量增长,约4200万t。对废旧橡胶轮胎进行掩埋或焚烧处理是简单而有效的方法,但极易引起空气和水污染等环境污染问题,不能从根本上解决废旧轮胎的处理问题。因此,大力发展废旧轮胎综合利用产业,对我国乃至全世界来说都意义重大。
化学法回收废旧橡胶生产再生胶,是我国目前采用的主要方法。但是,化学法回收生产过程易产生二次环境污染,因此需要开发其他回收方法。目前,其他的橡胶回收方法,如机械化学共同作用法、机械再生法、微波辐射法和超声波法等等制备再生胶工艺,由于生产成本高、效率低、技术难度较大等因素,尚未推广应用。因此,研究开发绿色高效的废旧橡胶回收的新方法、新工艺、新技术、新装置备受各国学者关注。
目前,一种利用双螺杆挤出机对废旧橡胶进行脱硫的新方法、新技术正引起越来越多的学者的关注。该方法最早是由加拿大TzoganakisC等和日本MasaakiK等学者报道的,常州大学的陶国良教授已利用此方法对脱硫工艺进行了较为系统的研究,取得了一些有益的成果。在脱硫实验技术不断改进的同时,关于橡胶脱硫机理的理论研究也取得了一定的进展。由于橡胶脱硫工艺不同,不同工艺脱硫机理也存在较大差异。目前已提出的有De-link法脱硫机理、常温脱硫机理、微波脱硫机理等,但尚不成熟。对于双螺杆挤出机脱硫,目前尚未见有相关脱硫机理的研究报道。由于双螺杆挤出机脱硫其实是一种“热力法”脱硫,通过导入外界的热能和机械能以选择性破坏交联键,所以,探讨脱硫机理必然要讨论含硫交联键的断裂能问题。
随着计算机技术、分子力场、分子模拟算法的不断发展,计算机模拟在材料领域的发展已经进入一个崭新的阶段,成为区别实验研究与理论研究的第3种研究方法。分子动力学模拟更是成为联系物质微观信息与宏观性质的基本方法,可以提供实验过程中无法获得或很难获得的信息。汪敬等应用从头算分子动力学模拟方法以及密度泛函理论对5-硝基-1-氢-四唑衍生物的热解机理进行了研究。报道了3条相关的反应途径,包括直接开环途径和质子转移途径。江德正等基于Amber力场结合量子力学对生物质主要组分纤维素热解过程进行了分子动力学模拟。利用半经验方法对不同聚合度的纤维素链进行了优化,得到最低能量的优化结构。分子动力学方法模拟得到纤维素分子链在加热过程中的断键顺序以及一次热解的基团,并分析了一次产物。殷开梁教授曾应用分子动力学模拟手段对正癸烷的热裂解进行了初步研究。提出了CHEN-YIN修正力场,并应用该力场对简化后的一种气态和两种液态的正癸烷系统的热裂解进行了分子动力学模拟。可以看出应用计算机模拟手段研究化学键断裂过程是可行的,但分子动力学模拟方法应用于脱硫机理的研究,尚未见文献报道。
本文将从分析化学键能的角度,通过将实验数据、理论模型和计算相结合的方式,初步探讨橡胶脱硫机理中的能量问题。这种新的研究方法,对探索硫化橡胶脱硫过程及脱硫机理,指导具体实验的顺利进行,具有重要的现实意义。
1模拟实验部分
1.1双螺杆脱硫原理
根据硫化橡胶主链C-C键和含硫键的键能差异,通过控制双螺杆挤出机提供的热能和剪切能大小以及其他加工参数,对硫化橡胶中低能化学键如单硫键、双硫键和多硫键进行选择性地破坏,减少对橡胶主链C-C键的破坏,以使硫化橡胶达到适当程度的脱硫,继而可经再加工获得较高性能的再生胶。
1.2模型构建与模拟细节
以美国Accelrys公司的MaterialsStudio4.0软件为平台,利用Discover模块进行能量最小化和分子动力学模拟实验。利用聚合物及分子建模工具构建体系A1、B1;根据实际橡胶密度,构建与体系大小相对应的立方P1晶胞体系A2、B2、C、D。力场采用COMPASS力场,模拟采用NVT不变的正则系综,温度控制采用Andersen方法,积分步长为1fs。每个体系先进行结构优化(最小化),以避免高能原子重叠。然后在400K下进行足够时长的预平衡,取出最后一个构象,将其链间交联键断裂,这样就得到交联键断裂前后的两种模型体系;对这两种体系继续进行预平衡与动力学模拟。由于体系的大小与结构不同,所以设定的模拟时间也不相同(表1),动力学模拟时每隔50fs收集1次轨迹。
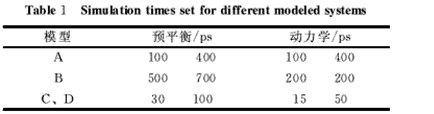
表1 不同模型体系设定的模拟时间
本工作共构建了4种橡胶模型。①模型A:2条链天然橡胶NR模型。以异戊二烯为重复单元,20个重复单元构成NR链;2条NR链通过数个单硫或多硫键Sn(n=1-3)相连,为体系A1(分子式C204H330Sn);A1的周期性结构为体系A2。②模型B:2条链丁苯橡胶SBR模型。SBR是苯乙烯与1,4-丁二烯和1,2-丁二烯的3单元共聚物,根据丁苯橡胶中各个单元的实际含量,估算出n(苯乙烯)∶n(1,4-丁二烯)∶n(1,2-丁二烯)为3∶8∶2;以此比例为依据,按不同的单元连接次序构建了2条链的SBR体系B1,链间通过含硫键相连接(分子式为C388H514Sn),体系B2为B1的周期性结构。③模型C:8条链天然橡胶NR周期模型。为增加模型中样本的数目,构建了8条30个重复单元的NR链,每条链含有2个交联键,其周期结构为体系C(分子式为C1200H1904Sn)。④模型D:8条链丁苯橡胶SBR周期模型。以2条链的SBR模型为基础,构建了8条3个重复单元的SBR链,每条链含有2个交联键,其周期结构为体系D(分子式为C1584H2096Sn)。4种模型6个体系结构示意见图1。
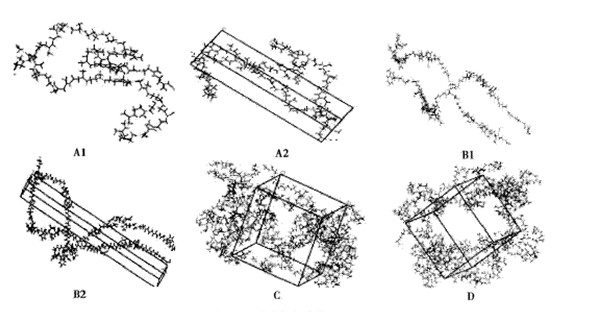
图1 6种硫化橡胶模型
2模拟结果
2.1计算原理
研究所需的含硫键单键键能早已有成熟的数据,对应于含硫单键断裂前后的能量差。而实际体系中,键的断裂过程的能量变化不仅涉及到单键键能的变化,同时还伴随着链分子构型能、片段间相互左右能、体系运动动能等诸多能量变化,所以在探讨能量变化时需考虑更多的因素。同时,按动力学的原理,键的断裂必经历键的活化过程,外界所需提供的能量应对应于活化能,而非断裂前后的能量差。但是,计算活化能需研究过渡态,这是一个非常棘手的课题,目前尚无简单有效的研究方法。本课题所采用的MS软件可以较为准确地计算较大体系如聚合物体系在一定温度压力下的时间平均能量,由分子动力学模拟计算出的同一温度压力条件下体系在含硫键断裂前后的平均能量差,可以粗略反映出含硫键的断裂难易程度,对应为断键外界所需提供的最小能量。
2.2结果与讨论
硫化橡胶具有三维立体交联结构,交联结构由主链C-C键、侧链单硫键C-S-C、双硫键C-S-S-C和多硫键C-S-S-S-C组成,4种键键能(长破折号)大小依次为:93、50-60、35和27kcal/mol,其中,多硫键的键能在橡胶所有化学键里是最低的[32]。通过对断键前后的系统进行动力学模拟,可以得出一系列的能量差数据,这些数据能在一定程度上反映C-S键,S-S键,S-S-S键的断裂难易程度,可以据此从化学键能的角度解释脱硫机理。
2.2.1模型A和B
模型A为2条链的NR模型,A1结构相对比较平滑,主链结构比较清晰,不具有长的支链,链之间的空间位阻较小,能量波动相对不会太大;周期性模型A2能够更好地模拟交联键在实际硫化橡胶中的环境,应该比A1更能准确地反映出系统真实能量的变化。根据模拟结果进行计算,得到的含硫键断裂前后系统的能量差结果示于表2中,并对不同模拟时长的计算结果进行对比。
从表2可以看出:①NR模型体系含硫键断裂前后的能量差均为正值,合理;②从数值上看,虽然能量差数值与相应含硫键键能间有一定的差别,普遍偏小,但是还是存在较好的定性关系,特别是二硫键和多硫键间;③3种情况下特别是周期体系S-S键断裂前后系统的能量差均比C-S键的大,不合理,这可能与C-S键断裂后对主链结构影响较大有关;④当预平衡和动力学模拟时间均从100ps增加到400ps时,能量差数据似乎更为合理,说明增加预平衡和模拟时间确实有助于提高计算结果的可信性;⑤对模型通过周期性排列改善后,再增加模拟时间,获得的能量差数据相对较为合理。
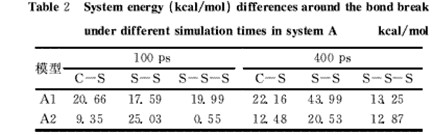
表2不同模拟时长模型A体系键断裂前后能量差
对模型B的2种SBR模型,采用了更长的预平衡时间和动力学模拟时间,其能量差模拟结果汇总在表3中。
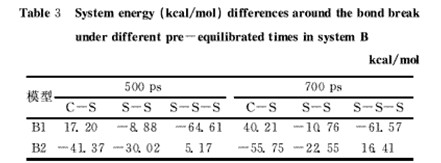
表3 不同预平衡时长模型B体系键断裂前后能量差
表3的数据可以看出,尽管模拟时间增加不少,但B1模型中除C-S键外,二硫或多硫键键断裂前后的能量差出现了负值,B2模型也仅S-S-S为正值,这很不合理,可以解释为:由于链的柔性及复杂性(三单元混聚),键断裂后,随着模拟时间的增长,链的结构及相对位置发生了较大改变,非键能的影响超过了断裂的键能,从而使能量差出现负值。显然,这样的模型不适合应用于本文所涉及的能量差计算。
2.2.2模型C
通过对模型A的研究和结果分析,考虑构建了较为复杂的且更为接近真实天然橡胶的模型C。C中每根链与其它链均生成2条交联键,从而构成类似于天然橡胶的交联网络结构;每个模型共有8个交联键;交联键均为单硫键的命名为S1,均为二硫键的命名为S2,三硫键的命名为S3。由于系统较大(含3000多个原子),计算速度较慢,所以预平衡时间和动力学模拟时间设定得较短,分别为30和15ps、100ps和50ps。研究断裂前后能量差时,采取随机断裂8个交联键中的1、2、3、4、8个键的方式。对键断裂前后系统的能量差进行计算,折算成单键断裂的数值,结果列于表4中。
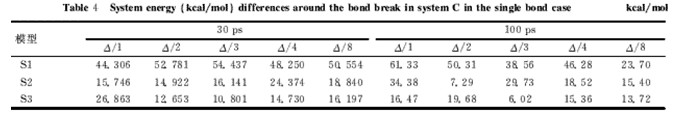
表4 不同模型C体系平均每键断裂前后能量差
从表4的数据可以看出:①能量差均为正值,合理;②除个别键断裂数据异常外,其它情况下C模型中平均单个键的能量差数据波动幅度不大,很好地解决了A、B模型中出现的数据波动较大的问题;③当预平衡和动力学模拟时间分别从30ps、15ps增加到100ps、50ps时,能量差数据也较为合理,但总体偏小,这说明增加预平衡和模拟时间确实对计算结果存在影响,数据稳定性尚需提高;④计算的能量差不完全对应单键能,这在前面的讨论中已作解释,而表4中数据显示,多数键断裂能量差特别是30ps的数值与单键能存在仅约10kcal/mol左右的偏差,结果非常理想;⑤能量差大小:单硫键>二硫键>三硫键,这与键能理论值顺序完全一致,说明C模型非常适合于研究含硫键的断裂能量差。
2.2.3模型D
对应模型B,构建了8条链SBR周期模型D,采用了模型C同样的模拟时间和模拟方法,其模拟结果如表5所示。
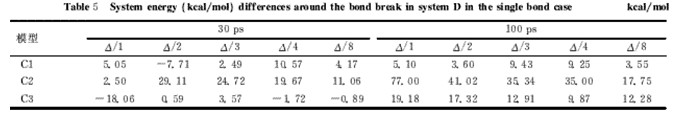
表5 不同模型D体系平均每键断裂前后能量差
从表5的数据可以看出,①30ps的能量数据出现不少负值,不合理,主要原因是SBR链的复杂性导至非键能的影响超过了断裂键能,而100ps的能量差均为正值,合理。②当预平衡和动力学模拟时间从30ps、15ps增加到100ps、50ps时,能量差数据也更为合理,这说明增加预平衡和模拟时间确实有助于提高计算结果准确性;③就100ps数据而言,单硫键断裂能的数据偏差较大,双硫键能量数据波动很大,仅三硫键断裂能偏差和波动都比较小,结果不理想;④能量差大小:单硫键<二硫键>三硫键,这与键能理论值顺序不一致。所以D模型不适于用于研究含硫键的断裂能量差。
4结论
分别构建了含2条链的天然橡胶(NR)、丁苯橡胶(SBR)以及8条链的NR、SBR周期模型,利用分子动力学方法对几种模型的能量进行了研究。通过对这些模型进行分子动力学模拟计算,得到了各模型的能量差数据。计算结果表明,原子数较少的2条链的NR模型得到的数据有较好的定性关系,较为合理。相对而言,SBR模型的能量值不太稳定,且出现负值,尤其二硫键的数据波动较大,这样的模型不适合应用于本文所涉及的能量差计算。而8条链NR周期模型系统的能量值则较为稳定,键断裂能量数据波动不大,且与理论值偏差较小,数值基本合理。实验表明,可以采用8条链NR周期模型来模拟硫化橡胶脱硫。此硫化橡胶模型与废旧轮胎橡胶的化学环境相似,但是实际的废旧轮胎橡胶中单硫、双硫和多硫键是按照一定的比例存在于三维立体交联结构中的。因此,需在后续实验中,将模型进行改进,尽量使其与实际的化学环境接近,以期得到更为精确的键能数据,从而为后期研究热力法脱硫机理提供合理的模型。