刘富,左丹英,操建华,朱宝库,徐又一
(浙江大学高分子科学研究所,浙江杭州310027)
Development of composite membranes of organic/ inorganic support
LIU Fu, ZUO Dan-ying, CAO Jian-hua, ZHU Bao-ku, XU You-yi
(Institute of Polymer Science, Zhejiang University, Hangzhou 310027, China)
Abstract:This paper gives a review on recent research and development of composite membranes of organic/ inorganic support. Preparation and application are introduced based on formation of organic membrane and interphase interaction between organic and inorganic support. Moreover its potential development is discussed.
Key words:composite;preparation;interphase interaction;solution deposit;grafting polymerization
摘要:从有机膜形成方式、有机膜与无机膜界面相互作用等方面对复合膜的制备技术和应用进行了的综述,并讨论了其潜在的发展和应用。
关键词:复合膜;制备技术;界面相互作用;溶液沉淀法;接枝聚合法
中图分类号:TQ028. 8;TB332 文献标识码:A
文章编号:1001-9731(2004)增刊
1 引言
高分子膜具有优良的分离性能、柔韧性好、制作简单、单位装置的膜使用面积大、品种多等优点,从而大规模应用于水处理、化工、生物、医药等领域。但聚合物膜存在不耐高温、抗腐蚀性差、机械强度不好、化学稳定性差等缺点,并且易堵塞,不易清洗[1]。无机膜,则具有许多独特的性质,机械强度高、热稳定性好、耐化学和生物侵蚀、使用寿命长,并且易于消毒和清洗。但是无机膜的不足之处在于抗污染能力差,分离选择性差,而且陶瓷膜大多数由无机氧化物制得,因而不能在碱性条件下使用[2,3]。为充分利用有机和无机膜的优点,克服各自的缺点,制备有机-无机复合膜已经成为膜研究的一个热点。复合膜按结构可以分为三种:无机物填充聚合物膜[4, 5];无机/有机杂化膜[6~8];有机物填充无机膜,也称为有机/无机支撑复合膜[9~12]。前两种膜主要是用溶胶-凝胶方法制备。本文在大量收集国内外此领域相关文献的基础上,对第三种复合膜的制备和应用进行归纳和总结。
2 聚合物溶液沉淀相转化法
沉淀法主要是溶剂蒸发沉淀相转化法,将聚合物溶液刮涂于多孔的或无孔的无机支撑物上,使溶剂蒸发,得到了均匀致密的聚合物膜皮层[2]。根据需要,可以反复进行以上过程。除了刮涂外,还可以根据无机支撑膜是管式的还是板式而采取浸涂、喷涂或旋转涂敷。此复合膜有机相与无机相之间主要为物理相互作用,聚合物膜厚范围为0.1~50µm。无机支撑膜为陶瓷或玻璃膜。除了溶剂蒸发沉淀相转化法外,也有人利用浸没沉淀相转化法制备了致密的聚偏氟乙烯/陶瓷复合膜[13]。以上复合膜主要用于气体分离、全蒸发和反渗透,也有人用于了微滤[9]和超滤[10, 14]。溶剂蒸发沉淀制备复合膜应注意以下几个问题。首先要注意孔渗现象。由于采用多孔无机膜做支撑体,毛细管力的作用使在涂膜过程中发生孔渗,使无机支撑膜的传质阻力增大。可以用不同的方法来避免或减少孔渗现象。最通用的方法是将无机膜孔预先填入某种物质,再涂膜,待有机膜形成之后,除去孔中的物质。如Matsumoto制备磺化聚砜/陶瓷复合膜时使用的填孔物质是壳聚糖,利用的壳聚糖在pH值小于6时可溶的、pH值大于6时变成悬浮液的性质[7]。另一种方法是采用高分子量聚合物并选用良溶剂,因为二者可使膜液中线团的流体力学半径增大。表1中列出了一些常用聚合物的溶剂和浓度。影响孔渗的另一个重要因素是无机膜的孔径分布,孔径分布应当越窄越好,此外表面孔隙率应尽可能高,一般情况下,空隙率为50%。
第二个要注意的是溶剂蒸发速率。特别是对于玻璃态涂层,快速的蒸发将会导致无机膜中产生应力,最终使得上面有机涂层产生裂缝。鉴于此,可以将溶剂的蒸发速度控制在一个较慢的水平上。Rezac等制备玻璃态芳香聚酰亚胺/陶瓷膜和聚碳酸酯/陶瓷膜时。将浸涂了溶液的无机物膜置于溶剂蒸汽饱和的环境中,通过控制环境中溶剂蒸汽的浓度达到控制涂层中溶剂蒸发速度[22, 23]。这样一个过程允许涂层可以缓慢地从一个溶胀的伸展状态向最终复合膜中平衡的互相贯通的状态转变。
第三个就是无机膜的表面改性问题。这里有两种方法进行表面改性。第一种方法是溶胶-凝胶改性,溶胶-凝胶方法是陶瓷等无机膜常用的修饰方法[3, 26]。
改性之后再进行有机膜的涂层[24, 25]。图1是改性无机上涂层有机膜形成的复合膜的结构示意图。第二种方法是热分解法,分解法是制备多孔SiO2、碳分子筛膜等的常用方法。一般是将硅橡胶涂敷在无机支撑体上,一定温度下热解,去除有机成分,涂层硅橡胶就形成以碳-碳、硅-氧等主链为骨架的无机网状孔结构。这种改性方法适用于空隙率很高或聚合物是玻璃态的情况。对聚酰亚胺溶液浸涂在两种方法修饰的无机膜上所制备的复合膜,进行了气体渗透实验,结果发现热分解法处理的复合膜的渗透率比用溶胶SiO2处理的要低50%。可以说,溶胶-凝胶比热分解法更有发展前途[23]。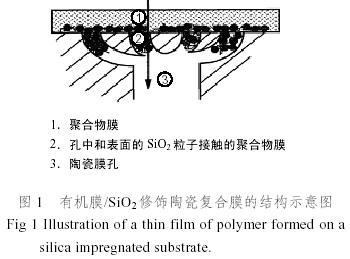
3 表面聚合法
通过化学方法使聚合物复合在无机支撑膜的表面或孔中。一是直接在无机膜表面进行单体的共聚或均聚,无机膜和聚合物膜之间是物理相互作用;二是对无机膜表面进行改性,使无机膜表面具有活性部位,然后通过活性部位进行单体的接枝聚合,这里无机膜和聚合物膜之间是通过化学键相互连接的。
3.1 表面原位聚合
3.1.1 气相聚合
Li在多孔玻璃上通过气相聚合反应制备了硅氧烷(SP)/无机复合膜。所使用的单体是二氯二甲基硅烷和二氯甲基乙烯硅烷,催化剂为NH3或(NH2)CO3.H2O。SP /玻璃复合膜进行气体渗透实验,实验结果为:对于溶解性小的气体,如He、H2、N2、CO、O2、Ar,它们的渗透速率随温度的升高而增大;另一方面,对于高溶解性的气体,如C2H4、CO2,它们的渗透速率随温度的升高而下降[27]。
3.1.2 溶液聚合
制备复合膜的有意义的一种有机物材料之一就是凝胶。凝胶一个显著的性质是当凝胶内部的环境如温度、pH、离子组成、溶剂组成或电场发生变化时,在凝胶中相应的会发生可逆的体积变化,正是这种可逆的凝胶内部体积的变化使得凝胶膜对有机溶剂/水混合物进行有效分离[28~30]。凝胶的机械强度太抵无法自身形成分离膜,必须将它复合在支撑膜上。Sakohara制备了聚丙烯酰胺凝胶(PAm)/陶瓷复合膜[28],将清洗后的陶瓷膜浸入过硫酸铵引发剂溶液中,取出后用蒸馏水清洗掉陶瓷膜外壁的过硫酸铵;接着将膜迅速浸入温度为70℃的丙烯酰胺、N, N’-亚甲基-双丙烯酰胺和N, N, N’, N’-四甲基乙烯基二胺交联剂的混合溶液中,N2保护下反应进行2h,从溶液中取出膜,在沸水中浸泡2h,即制得PAA/陶瓷复合膜。将此膜用于丙酮/水的全蒸发实验,得到了相当大的水通量,并且当原料中丙酮的含量达到95 mol%时,此膜的分离系数高达2000,如此高的分离系数主要是因为随着原料中丙酮含量的增加PAA的网络结构发生了收缩,收缩的网络结构阻拦丙酮分子的渗透。
3.1.3 电化学和光化学合成法
这两种方法都是在微孔无机膜上复合上一层超薄聚合物膜。Liu用电化学方法研究二乙烯基(DVB)和乙基乙烯基苯(EVB)在陶瓷膜表面的聚合反应[31],这两种单体在电解质溶液中经电化学还原成阴离子,然后通过阴离子聚合反应机理在修饰的陶瓷膜表面聚合生成DVB- EVB共聚物。用此膜进行气体传输实验、计时电流、计时电压实验,结果都证实所制得的复合膜为具有选择性的无缺陷膜。在光化学合成法中单体进行的是自由基聚合,因此通过这种方法制备复合膜的有机单体很多。表2中列举了一些通过光化学合成法在无机支撑膜上制备的膜的组成以及它们的单体。用表2中Ⅴ号乙烯基二茂铁-二乙烯基苯共聚物/Al2O3陶瓷膜进行气体渗透实验,O2/N2的分离系数是8.0,膜厚为40~3200 nm[32]。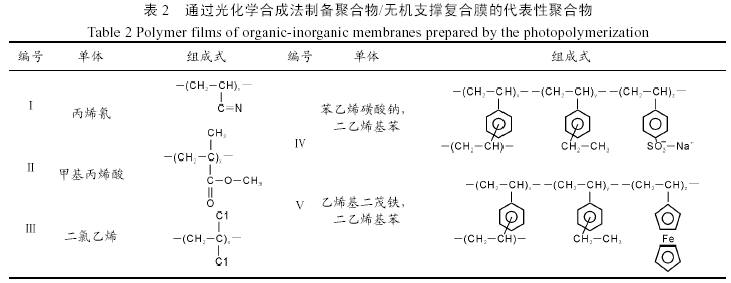
3.2 表面接枝聚合
3.2.1 纳米技术接枝法
原理是:以多孔无机膜为基体,选用有两个活性基团的化合物(M-R0-X)对此膜进行修饰,M基团与膜表面的OH等基团反应,修饰后的膜表面结构为:膜- R0-X。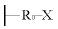
选择两种化合物X1-R1-X1和X2-R2-X2作为第一、二共聚单体(X1,X2都是活性基团,且X,X1,X2能相互反应,X 和X2可以是相同的)。第一步:将单体一与上述修饰过的陶瓷膜表面接触,X1与膜表面的X基团进行反应,生成以下结构:
将残存的单体一清洗掉。第二步:X2-R2-X2与膜接触反应,生成以下结构: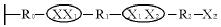 将残余的单体二清洗掉,重复上述两个步骤,直到得到需要的聚合度,结构如下:
将残余的单体二清洗掉,重复上述两个步骤,直到得到需要的聚合度,结构如下: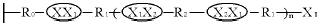
还可在共聚到一定程度时,引入第三、四种单体X3-R3-X3和X4-R4-X4与膜接触反应。控制重复反应的次数,从理论上来说就可以控制结合在陶瓷膜表面和膜孔壁上聚合物链的长度,从而控制复合膜的孔径。所以这种方法在理论研究和实际应用上都很有意义。
Okazaki和Sawamoto等人用(C2H5O)3Si- CH2CH2CH2NH2(3-氨基丙基三乙氧基硅烷)对多孔陶瓷膜进行表面修饰,使无机膜表面带有-NH[33, 34]2。Okazak采用的是液相纳米技术接枝过程,酸酐和二胺的溶液分别与修饰的陶瓷膜接触,生成表层有聚酰亚胺(PI)复合膜。酸酐和二胺在-NH2修饰的陶瓷膜上重复反应的过程如图2。重复反应次数不同,PI /陶瓷复合膜对CO2/CH4的分离系数的变化范围为1.0~6.4,分子截留范围为400~4000。采用液相纳米技术接枝过程,每步反应之后的残余单体没有清洗干净,这些单体在以后的反应中又参与了反应,致使膜孔被堵。鉴于残余单体难于清洗,Sawamoto改用气相纳米接枝过程,反应原料和步骤类似,但各反应物都是以气态形式与陶瓷膜接触,得到的PI /陶瓷复合膜对CO2/CH4的分离系数的变化范围为0.87~16[34]。
3.2.2 溶液接枝聚合
这种方法也是先对无机膜表面进行修饰,使表面带有某种官能团(活性点),然后再引进单体聚合,这样,有机膜和无机膜之间是共价键相连。改性剂一般用硅烷RSiX3,X是可水解的基团,如卤素、氨基、烷氧基和酰基,R是不可水解的基团,是无机膜表面进行后续接枝聚合的活性点。
Castro在修饰的多孔SiO2膜表面进行了1-乙烯基2-吡咯烷酮(VP)接枝聚合,制备了聚乙烯吡咯烷酮(PVP)/SiO2复合膜[35]。选择的改性剂是乙烯基三乙氧基硅烷(VTMS,CH2=CHSi(OCH2CH3)3),在二甲苯中它与SiO2膜表面的-OH发生水解反应,生成了表面带有乙烯基活性点的SiO2膜,SiO2膜表面修饰的过程如下式: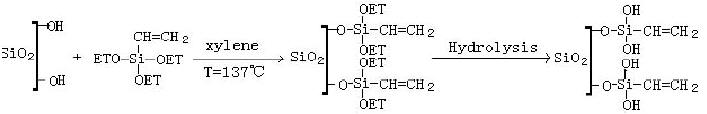
Chaimberg等人对VP溶液浓度、反应温度、时间及接枝率进行了研究,确定了最佳的反应工艺[36, 37]。将SiO2膜和PVP/SiO2复合膜进行水、乙醇、丙醇、丁醇、甲苯、环己烷的渗透实验,结果是PVP/SiO2复合膜中6种溶剂的渗透率大大减小,减小率分别为47.8%、40.4%、45%、52.3%、24.0%、14.0%,很明显,具有极性的脂肪醇和水的减小率大于无极性的甲苯和环己烷,除了SiO2膜孔被PVP堵塞这一原因外,主要还是因为在良溶剂水、乙醇、丙醇、丁醇中,接枝PVP链舒展导致PVP接枝膜溶胀,孔径减小,随氯代2-甲基乙烯基硅烷对多孔玻璃表面进行修饰,Otake等人所使用的聚合单体是酰胺(Am)和丙烯酸(AA),Tsuji所使用的单体是N-异丙基丙烯酰胺(NIPAm),两组实验中,单体之间的共聚是自由基聚合机理。制备复合膜的过程与Sakohara相同[28]。以上两种复合膜也进行丙酮/水以及聚乙烯醇/水/丙酮的分离实验。结果显示,随着原料中丙酮含量的增加,Am-AA共聚物凝胶层和IPAm均聚物凝胶层网络结构都进行一定的收缩。使得丙酮的截留率增加。此结果与Sakohara的实验结果相同,即溶液中溶剂的浓度变化引起的聚合物膜的溶胀和收缩直接影响到膜的渗透率和分离因子。Yoshida也在VTMS修饰的陶瓷膜上接枝醋酸乙烯酯(VAc)-乙烯吡咯烷酮(VP)共聚物,此复合膜用于甲醇和甲基叔丁基醚的全蒸发分离实验,原始液中甲醇浓度在1%~5%(v/v)变化时,膜分离因子的范围为26~100[38]。Jou也用引发剂接枝聚合方法在管式SiO2膜上接枝聚醋酸乙烯酯(PVA),此复合膜从水中脱除氯仿和三氯乙烯的脱除率达到69%~106%[39]。
3.2.3 等离子体接枝聚合法
等离子体聚合是一种较新的聚合方法,无机多孔膜等离子体处理后,一定条件下再与聚合单体接触,单体就会在其表面和孔壁上接枝聚合,生成线性聚合物,聚合反应可以通过在反应管中引入空气终止,最终可得到接枝程度不同的复合膜[40],复合膜中聚合物层与无机膜层之间结合得非常紧密。如Kai等人[41]以多孔玻璃膜为基体,丙烯酸甲酯(MA)为单体,采取直接等离子接枝和两步等离子接枝的方法制备复合膜(图3)。前一种方法单体直接在等离子体处理过的玻璃膜孔壁上接枝聚合,所形成的层厚约50μm;后一种方法分两步,第一步以单体和交联剂的混合物为原料,在多孔玻璃膜孔表面接枝聚合一簿层交联聚合物,第二步是在这一层交联聚合物上,单体聚合形成线形接枝聚合物,这种方法得到的接枝层厚为25~30μm。这两种方法制备的聚丙烯酸甲酯/多孔玻璃复合膜在氯仿/环己烷混合物的全蒸发和蒸汽渗透实验中对氯仿有很高的选择性,28%的环己烷的氯仿溶液的渗透率达到0.5kg/m2 h,对氯仿的分离因子高达16.3。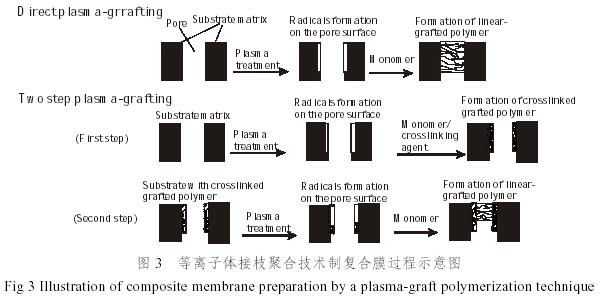
4 表面有机物化学改性
4.1 表面有机物单分子层接枝
这种方法所用的无机膜孔径在4~10nm之间,原理是:在无机膜表面通过化学共价键接枝上一超薄有机物分子膜,这样的有机物分子有有机膦化物[42~44]、氯硅烷衍生物[45, 46]、硅氧烷衍生物[47]等等。这些有机化合物很容易与SiO2、Al2O3、ZrO2、TiO2、玻璃等无机膜表面上的-OH基团反应,生成M-O-Si键。
Randon在TiO2、ZrO2膜表面进行烷基磷酸和烷基膦酸接枝反应[42],烷基磷酸和烷基膦酸衍生物接枝层大大改善了无机膜表面与溶液中离子的相互作用,使得无机膜表面极性亲水性变为非极性疏水性。用烷基磷酸和烷基膦酸/ ZrO2复合膜进行血牛清白蛋白(BAS)过滤实验,当溶液的pH=7时,通量和截留率都增加了。接着Randon在孔径为5nm的γ- Al2O3陶瓷膜上接枝了n-丁基膦酸(n-H4H9PO3H2)和n-十二烷基磷酸盐(n-C12H25OPO3H2)分子层[44],此复合膜进行气体渗透实验,发现CH4、C2H6、C3H6和CO2的透过率较高,C3H8/N2的分离比高达10,其分离效果比PE膜好。
Miller在平均膜孔径为5nm的γ- Al2O3陶瓷膜表面及孔中接枝硅烷十三氟-1,1,2,2-四氢辛基-1-三氯硅烷(TDFS)单分子层[45]。由于位致障碍并非所有的羟基都发生反应,于是TDFS基团之间留下了孔隙,这些孔隙对气体或液体的透过性与选择性均有影响。TDFS/γ- Al2O3陶瓷复合膜对甲苯和润滑油有较大的分离比。Leger也进行了三氯十八硅烷在平均膜孔为5nm 的Al2O3陶瓷膜表面的接枝反应[46],用此膜进行气体渗透实验,结果发现所有气体的渗透率比没有复合前下降了三个数量级,而且此复合膜不适宜做这些气体的分离膜。Leger还以PDMS为表面接枝物、多孔Al2O3膜为支撑物制备了PDMS / Al2O3复合膜[47]。纯水不能渗透此复合膜,将此膜用于从水溶液中抽提有机溶剂的全蒸发实验,结果发现,有机溶剂的渗透通量很高,这说明硅氧烷/陶瓷复合膜能应用于挥发性有机化合物的抽提。PDMS有机膜层增加了陶瓷膜表面的疏水性,而且没有堵孔现象发生。
4.2 表面有机物吸附
利用无机膜表面的吸附活性中心OH基团,进行有机物的化学吸附以制备有机/无机复合膜。Dafinov工作组[48]通过Al2O3膜表面化学吸附醇进行了醇/Al2O3复合膜的制备,醇与Al2O3膜之间的化学吸附生成Al-O-R共价键。此复合膜在200℃以下使用都是稳定的,没有发现分解脱附现象,吸附的醇使Al2O3膜表面的亲水性大大降低,结果水通量大大减小。
5 部分热解法
聚合物的热解即是它在高温环境下发生的降解过程。完全热解时,聚合物中所有不稳定的共价健都会断裂,不稳定的元素都被蒸发移走,一般得到的是脆性产品。如果样品在高温条件下放置的时间太短或是温度偏低,它只能部分热解,生成物中还会有大量有机成分存留下来,它既保留了部分弹性,热稳定性又大大提高.并且调节热解温度,可以控制产品中有机与无机组分的比例,从而调节其性能。此方法制备的复合膜中有机层的厚度为(30±5)μm。Shelekhin等人将PSS ( Polysilastyrene, dimethylsilane-metylphenylsi- lane co-polymer) 溶液涂敷在一微孔玻璃膜上,经紫外辐照,PSS交联而成的膜,再于382~470℃氮气氛中热解,得到的膜元素组成C/Si、H/Si、H/C都介于PSS与SiC(PSS完全热解产物)之间[49, 50]。用此复合膜进行H2/SF2的分离实验,发现此膜同时具有膜和分子筛的气体传输的特性,热解温度为470℃时,H2/SF2的分离系数达到328,而没有进行热解的复合膜的分离系数只有10。Stevens等人[51]也利用部分热解法制备PDMS/玻璃复合膜,他们将涂敷的PDMS部分热解与氧化交联结合起来,部分热解后的PDMS发生交联反应。
6 总结与展望
根据以上的总结,我们可以从以下两个方面来探讨聚合物/无机支撑复合膜的新的制备方法。首先,在以上各种复合膜中,聚合物层与无机层之间的粘合是物理相互作用或共价键相互作用,其实在层间还可以有另外一种相互作用:非共价键相互作用(如静电相互作用、氢键相互作用、电荷转移等)[52, 53],所以制备聚合物/无机复合膜的一个可以探讨的方法就是自组装膜,此方法已经应用于聚合物填充聚合物膜[54,55];原理:对无机膜表面进行修饰,使其表面带将负电荷,将带负电荷的固体表面与溶液阳离子聚电解质接触,吸附,然后用水洗净,使表面正电,再浸入阴离子聚电解质中,取出,表面带负电,如此往复进行,即可形成多层聚合物自组装膜/无机复合膜。第二,从复合膜的用途方面着手,从前文可以看出,复合膜有机层是致密结构,使得这类膜主要应用于气体分离、全蒸发和反渗透;为了使这类膜能广泛用于微滤、超滤、膜生物反应器,就必须增大复合膜中聚合物膜的孔径;目前制备多孔膜的方法,最主要的是浸没沉淀相转化法。从理论上说,改变铸膜液的组成、凝固液的组成和温度以及对无机支撑物的表面修饰和改性都可以对聚合物膜的孔径进行控制。因此通过浸没沉淀相转化法制备多孔聚合物/无机支撑复合膜也是今后值得研究的一个方面。
参考文献:
[1] 刘茉娥, 等. 膜分离技术[M]. 化学工业出版社, 1998.
[2] Marcel Mulder著. 李琳译, 单德芳校, [M]. 膜技术基本原理, 第二版, 清华大学出版社, 1999.
[3] 韦奇, 王大伟, 张术根. [J]. 功能材料, 1999, 30(6): 601-603.
[4] 钟顺和, 李传峰, 孙宏伟, 等. [J]. 膜科学与技术, 2002, 22(4): 21-25.
[5] Bottino A, Capannelli G, Asti V D, et al. [J]. Separation and Purification Technology, 2001, 22-23: 269-275.
[6] Nomura S, Fujii T, Suzuki M. [J]. Water Science Technology, 1997, 35(8): 137-144.
[7] Matsumoto Y, Sudoh M, Suzuki Y. [J]. Journal of Membrane Science, 1999, 158: 55-62.
[8] Hong Y K, Hong W H. [J]. Journal of Membrane Science. 1999, 159: 29-39.
[9] Liu Q L, Li Q B. [J]. Journal of Membrane Science. 2002, 202 : 89-95.
[10] Smaïhi M, Jermoumi T, Marignan J, et al. [J]. Journal of Membrane Science, 1996 116: 211-220.
[11] Zoppi R A, Neves S das, Nunesc S P. [J]. Polymer, 2001, 4:5461-5470.
[12] 苏静, 邓国宏, 余立新,等. [J]. 清华大学学报(自然科学版), 2001, 41(12): 35-37.
[13] Margaret K, Kishor P. [P]. EP 0674937A2.
[14] Matsumoto Y, Sudoh M, Suzuki Y.[J]. Journal of Membrane Science, 1999, 157: 139-144.
[15] Linkov V M, Bobrova L P, Timofeev S V, et al. [J]. Materials Letters, 1995, 24: 147-151.
[16] Zhu Y H, Chen H F. [J]. Journal of Membrane Science, 1998, 138:123-134.
[17] 陈光文, 袁权, 吴迪镛, 等. [J]. 化工学报, 2000, 51(6): 725-733.
[18] 陈光文, 袁权. [J]. 高校化学工程学报, 2001, 15(5): 420- 424.
[19] Chen W J, Charles P A, Martin R. [J]. Journal of Membrane Science, 1995,107:199-207.
[20] Aranda P, Chen W J, Martin C R. [J]. Journal of Membrane Science, 1995, 99: 185-195.
[21] Zhu Y, Minet R G, Tsotsis T T. [J]. Chemical Engineering Science, 1996, 51(17): 4103-4113.
[22] Rezac M E, Pfromm P H, Costello L M, et al. [J]. Ind. Eng. Chem. Res. 1993, 32, 1921-1926.
[23] Rezac M E, Koros W. [J]. Journal of Applied Polymer Science, 1992, 46: 1927-1938.
[24] Moaddeb M, Koros W J. [J]. Ind. Eng. Chem. Res. 1995, 34:263-1740.
[25] Moaddeb M, Koros W J. [J]. Journal of Membrane Science, 1996, 111:283-290.
[26] 吴翠明, 徐铜文, 杨伟华. [J]. 无机材料学报, 2002, 17(4): 842-648.
[27] Li D, Seok D R, Hwang S T. [J]. Journal of Science Membrane, 1988, 37: 267-275.
[28] Sakohara S, Muramoto F, Sakata T, et al. [J]. Journal of Chemical Engineering of Japan, 1990, 23(1): 40-45.
[29] Otake K, Tsuji T, Konno M, et al. [J]. Journal of Chemical Engineering of Japan, 1988, 21(4): 443-445.
[30] Tsuji T, Konno M, Saito S. [J]. Chemical Engineering of Japan, 1990, 23(4): 447-452.
[31] Liu C, Espenscheid M W, Chen W J, et al. [J]. J. Am. Chem. Soc., 1990,112:2458-2459.
[32] Liu C, Martin C R. [J]. Nature, 1991,352: 50-52.
[33] Okazaki I, Ohya H, Semenova S I, et al. [J]. Journal of Membrane Science, 1998, 141: 65-74.
[34] Sawamoto S, Ohya H, Yanase K, et al. [J]. Journal of Membrane Science, 2000, 174: 151-159.
[35] Castro R P, Cohen Y, Monbouquette H G. [J]. Journal of Membrane Science, 1993, 84: 151-160.
[36] Chaimberg M, Cohen Y. [J]. Ind. Eng. Chem. Res. 1991, 30: 2534-2542.
[37] Chaimberg M, Parnas R, Cohen Y. [J]. Journal of Applied Polymer Science, 1989, 37: 2921-2931.
[38] Yoshida W, Cohen Y. [J]. Journal of Membrane Science, 2003, 213: 145-157.
[39] Jou J D, Yoshida W, Cohen Y. [J]. Journal of Membrane Science, 1999, 162: 269-284.
[40] Yamaguchi T, Nakao S, Kimura S. [J]. Macromolecules, 1991, 24: 5522-5527.
[41] Kai T, Yamaguchi T, Nakao S. [J]. Ind. Eng. Chem. Res., 2000, 39: 3284-3290.
[42] Randon J, Blanc P, Paterson R. [J]. Journal of Membrane Science, 1995, 98: 119-129.
[43] Randon J, Paterson R. [J]. Journal of Membrane Science, 1997, 134:219-223.
[44] Randon J, Blanc P, Paterson R. [J]. Journal of Membrane Science, 1995, 98: 119-129.
[45] Miller J R, Koros W J. [J]. Separation Science Technology, 1990, 25: 1257.
[46] Leger C, Liva H D L, Paterson R. [J]. Journal of Membrane Science, 1996, 120:187-195.
[47] Leger C, Liva H D L, Paterson R. [J]. Journal of Membrane Science, 1996, 120:135-146.
[48] Dafinov A, Garcia-Valls R, Font J. [J]. Journal of Membrane Science, 2002, 196: 69-77.
[49] Shelekhin A B, Grosgoget E J, Hwang S T. [J]. Journal of Membrane Science, 1992, 66: 129-141.
[50] Grosgogeat E J, Fried J R, Jenkins R G, et al. [J]. Journal of Membrane Science, 1991, 57:237-255.
[51] Stevens N S M, Rezac M E. [J]. Chem. Eng. Sci., 1998, 53(9): 1699-1711.
[52] Yang X., Wu G F, Hu Y. H., et al. [J]. Int. J. Mass Spectrometry, 2000, 202: 55-68.
[53] Schalley C. [J]. Int. J. Mass spectrometry, 2000, 194: 11-39
[54] Stroeve P, Vaspuez V, Coelho M A N, et al. [J]. Thin Solid Films, 1996,284/285: 708-712.
[55] Ackern F V, Krasemann L, Tieke B. [J]. Thin Solid Film, 1998, 327-329: 762-766.
基金项目:国家863基金资助项目(2002AA061203)
作者简介:刘富(1980-),男,在读硕士,2002年于青岛科技大学获学士学位,现在浙江大学高分子科学与工程系,师承徐又一教授,从事膜材料与工艺的研究
论文来源:中国功能材料及其应用学术会议,2004年,9月12-16日
