研究背景
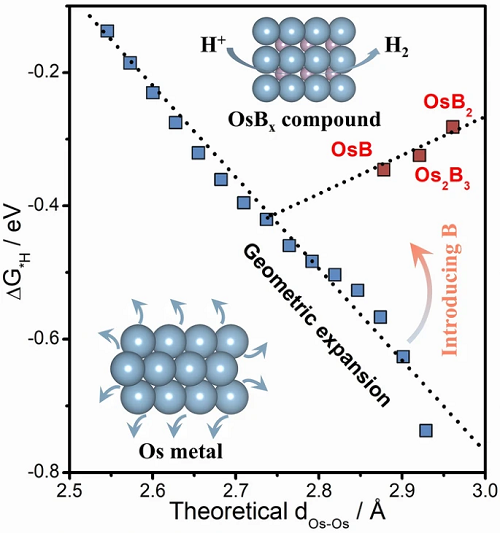
在原子尺度上精确调整金属活性中心间距对提高催化活性和加深对催化机理的认识具有重要意义,但仍然充满挑战。对此,武汉理工大学木士教授课题组利用轻原子稀释活性金属位点密度的方法实现了金属原子间距(dM-M)的精确可调,同时还发现了一种有别于常规的氢吸附模式。以活性金属锇(Os)和作为填隙原子的硼(B)为例,在OsBx金属间化合物结构中,如果逐渐增加B的含量,将会伴随着Os金属位点密度的减少,Os的dOs-Os将逐渐从2.73增加到2.96 A。然而,随着dOs-Os增加,氢的吸附-距离关系通过d带的下移而出现反转,打破了传统的认知,从而优化了催化过程中氢在电极表面的吸附和H2O的解离,最终使析氢反应(HER)活性几乎呈线性增加。在碱性介质中,具有最大dOs-Os值(2.96 A)的OsB2表现出最佳的HER活性(8 mV @ 10 mA cm-2),而且还抑制了O的吸附,从而提高了催化剂的稳定性。这种新型的原子级催化位点距离调制策略和氢的吸附-距离反转关系的发现为高效催化剂的设计提供了新的见解。
Tuning Active Metal Atomic Spacing by Filling of Light Atoms and Resulting Reversed Hydrogen Adsorption?Distance Relationship for Efficient Catalysis
Ding Chen*, Ruihu Lu*, Ruohan Yu, Hongyu Zhao, Dulan Wu, Youtao Yao, Kesong Yu, Jiawei Zhu, Pengxia Ji, Zonghua Pu, Zongkui Kou, Jun Yu, Jinsong Wu, Shichun Mu*
Nano-Micro Letters (2023)15: 168
https://doi.org/10.1007/s40820-023-01142-1
本文亮点
1. 密度泛函理论(DFT)计算表明,间隙B原子可以调整主金属Os的原子间距,并表现出反转的氢吸附-距离关系。
2. 建立了活性Os原子间距与催化活性之间的构效关系。
3. 在构建的多个OsBx物相中,具有最大B:O原子比的OsB2(dOs-Os=2.96 A)在碱性介质中表现出最佳的HER活性(8 mV @ 10 mA cm-2)和良好的电化学稳定性。
内容简介
利用间隙填充策略实现活性原子间距的精准调控及构效关系的解读具有重要科学意义,但该方面一直是研究的空白。武汉理工大学木士春课题组通过密度泛函理论(DFT)计算,首次证实了小半径和低电负性的硼(B)原子能够完美地平衡金属锇(Os)几何膨胀过程中的应力变化和电子转移;伴随着金属间化合物OsBx(x=1,1.5,2)中B间隙原子数量的增加,Os金属位点密度减少,Os原子间距(dOs-Os)从2.73逐渐增加到2.96 A,并获得了反转的氢吸附-距离关系,即间隙B原子填充在增加Os原子间距(dOs-Os)的同时亦削弱了Os对氢(H)的强吸附。与传统的表面修饰和掺杂策略不同,强的主客体电子相互作用和新化学键的形成进一步增强了催化剂的活性和稳定性。此外,通过研究还构建了OsBx的结构-活性关系,即活性Os原子间距随着B的逐渐填充而增大,导致H结合和H2O解离能垒减小;同时,增强的Os-B配位效应抑制了Os的失活和溶解,实现了催化剂活性和稳定性的同步提高。
图文导读
I 密度泛函理论(DFT)计算
理论计算结果如图1所示。图1a表明,OsBx金属间化合物随着B含量的增加,Os原子间距离dOs-Os从2.73逐渐增加到2.96 A,进一步导致空位(hollow site)的Os-H键长增加;图1b表明,与传统认识相反,具有小半径和低电负性的间隙B可以实现反转的氢吸附-距离关系;从图1c可看出,几何膨胀通常会导致Os-Os相互作用减弱,导致d带中心(εd)升高,吸附能力增强;而B间隙原子可以通过降低d带实现氢吸附-距离关系的反转。上述理论分析揭示了通过填充间隙原子B来梯度分散Os活性位点及提高HER活性是可行的,为后续数类似催化剂的合成和构效关系的确定指明了方向。

图1. 理论计算。(a)原子间距调制,紫色、蓝色和白色的球分别代表B、Os和H原子;(b) ΔG*H随Os-Os键长增加的变化,箭头表示引入B的效果;(c)B引入引起的d带位移示意图。
II 结构表征
如图2a表示,通过控制退火温度,热解得到三种金属间硼化物;图2b的XRD结果表明,在700℃、800℃和900℃下得到的产物分别为六方晶相OsB (OsB- H)和Os2B3 (Os2B3-H),正交晶相OsB2 (OsB2-O);图2c显示了Os、OsB、Os2B3和OsB2四个典型的Os L3边EXAFS谱图,表明填充间隙B原子诱导了新的Os-B键的形成以及dOs-Os逐渐增加; 图2d进一步描述了实验和理论得到的dOs-Os之间的线性拟合结果,证实了间隙B填充引起的Os原子的逐渐分散;图2e-h展示了沿Os [100],OsB-H [010],Os2B3-H[001]和OsB2-O[101]带轴的高分辨率晶格原子图像和相应的快速傅里叶变换(FFT)模式,与理论的晶体结构和原子排列非常吻合。以上结果充分证明了一系列有序金属间硼化物的合成,并实现了Os金属原子的可控分散。
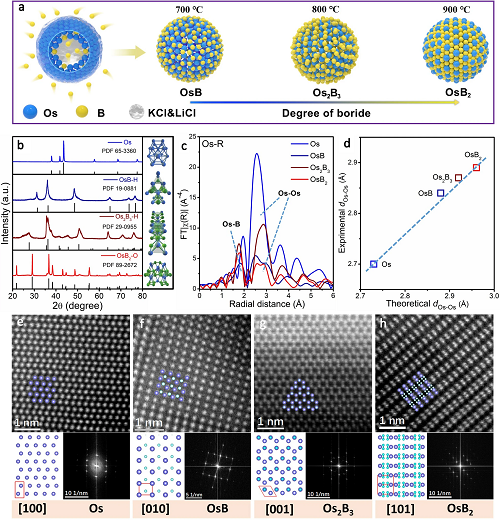
图2. 结构表征。(a)制备OsBx的示意图; (b)所有合成材料的XRD图谱; (c)Os、OsB、Os2B3和OsB2的L3 边EXAFS光谱; (d)理论与实验Os-Os距离的线性关系;(e-h)高分辨率HAADF-STEM图像及其相应的晶体结构和FFT图,其中Os原子呈蓝紫色,B原子呈蓝白色。
III 析氢反应(HER)活性
图3a-b表明OsBx在1 M KOH中的HER活性和动力学优于Os和商用Pt/C;图3c表示在过电位为50 mV时,OsB?具有最高的转换频率(TOF),分别是Os2B3和OsB的4.0倍和2.4倍;图3d显示了酸性介质中HER性能与ΔG*H之间良好的线性拟合关系,表明Os转化为OsB2导致的*H吸附减弱是促进HER活性的主要原因之一;图3e表示在Os、OsB、Os2B3和OsB2的Os位点上,H2O解离是速率决定步骤,限制了HER过程;图3f进一步对50 mA cm-2下的能垒和碱性HER活性进行拟合,良好的线性关系证明间隙B原子加速了H2O解离,从而提高了碱性HER活性;综上所述,B原子的引入减弱了*H吸附,加速了H2O解离。图3g比较了OsBx与最近报道的铂族金属电催化剂在1 M KOH条件下的HER性能,相比于大多数的Pt簇金属基催化剂,OsB2具有更好的HER活性。
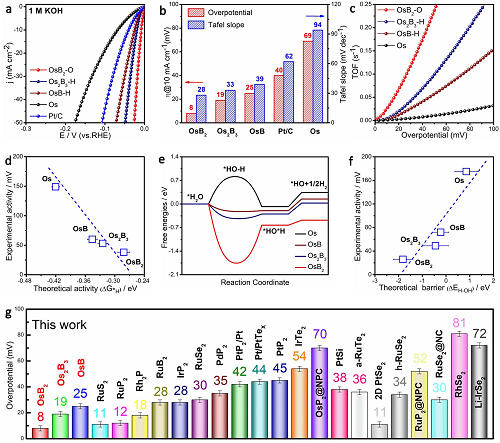
图3. HER活性。(a)HER极化曲线;(b)在10 mA cm-2下对应过电位和Tafel斜率;(c)TOF与Os和OsBx过电位之间的关系;(d)酸性条件下理论与实际活性的相关性;(e)碱性HER的自由能图;(f)碱性环境下H-OH分裂能垒与实际活性的关系;(g)比较OsBx与最近报道的铂族金属电催化剂在10 mA cm-2、1 M KOH条件下的HER性能。
IV 活性提高机理
图4a-b表明成功引入了间隙B;图4c的小波变换可视化了Os-B路径;图4d-e通过对比1500~1700 cm-1的原位拉曼光谱的宽峰,可以进一步证明OsB2相对于Os对H2O的吸附也明显减弱;图4f通过UPS探测Os和OsBx催化剂的占据电子态来了解B-O相互作用对氢吸附能力的影响,表明B和Os原子之间的p-d杂化导致Os的d带下移,使OsBx的εd发生负移;图4g显示了实验和理论εd之间良好的线性关系,进一步证明了εd是通过B-Os相互作用而下降的;图4h表明εd的下降减弱了Os 5d和H 1s之间的反应性,导致*H吸附较弱;图4i表示d带理论预测的这一趋势与计算得到的*H吸附也很吻合。

图4.活性提高机理。(a)对应产物B 1s的XPS谱图;(b)OsB?的L?边EXAFS拟合曲线;(c)EXAFS信号的WT;(d-e)Os和OsB2的原位拉曼分析;(f)Os、OsB、Os2B3和OsB2相对于费米能级的UPS价带谱;(g)实验与理论εd之间的校正;(h)*H s轨道与Os 5d轨道的相互作用;(i)理论εd、ΔG*H与HER活性的关系。
V 稳定性提高机理
最后,研究人员还探索了OsBx稳定性提高的机理。图5a表示催化剂在电解质中的活性降解可能是由于金属氧化态的形成导致了两条活性失效路径;图5b表示OsB、Os2B3和OsB2活性中心的Os原子表面εd分别为-1.24、-1.51和-1.91 eV。根据d波段理论,通过*O吸附而使εd上移会使反应性增强,从而导致*H吸附增强和HER性能下降,因此丰富的Os-B键抑制了O的吸附;图5c-d分别为不同加速循环后催化剂的过电位与εd的关系,以及电催化后OsB、Os2B3和OsB2溶解在电解液中Os的浓度。这些结果中OsB?的更优表现进一步证实了金属间硼化物结构越复杂,配位效应越强,在HER过程中抗氧化中毒和溶解稳定性越好。图5e进一步表示OsB2在酸性和碱性介质中耐久性试验(100 h)中都能保持稳定的工作电流。
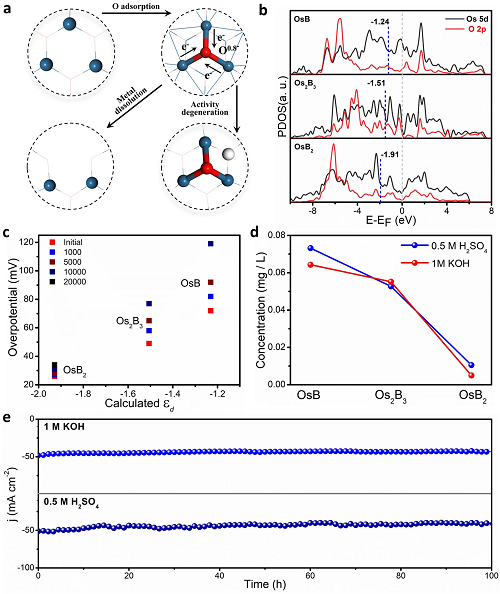
图5. 稳定性提高机理。(a)强O吸附诱导催化活性退化的两种可能机制;(b)相关氧原子和吸附氧的O2p轨道和O5d轨道的PDOS计算;(c)在1 M KOH中不同加速循环后催化剂的过电位与氧的关系;(d)电催化后OsB、Os2B3和OsB2溶解的电解质中Os的浓度;(e)OsB2在1M KOH和0.5 M H2SO4条件下的耐久性。
作者简介

陈钉
本文第一作者
武汉理工大学 博士研究生
▍主要研究领域
催化剂的设计合成与催化机制分析。

木士春
本文通讯作者
武汉理工大学 教授
▍主要研究领域
(1)燃料电池催化剂;(2)电解水产氢催化剂。
▍个人简介
武汉理工大学首席教授,博士生导师。长期致力于氢能源催化材料研究。以第一作者或通讯作者在Nat. Commun.、Adv. Mater.、J. Am. Chem. Soc.、Angew. Chem. Int. Ed.、Energy Environ. Sci.等国内外期刊上发表280余篇高质量学术论文。
▍Email:msc@whut.edu.cn
撰稿:原文作者
编辑:《纳微快报(英文)》编辑部