背景介绍
电催化水分解作为一种可持续氢气生产的前景技术,受到了广泛关注。然而,阳极的氧气析出反应(OER)存在反应动力学缓慢的问题,极大地限制了水分解的效率。因此,深入理解OER催化机制,对于设计高效OER催化剂具有重要意义。根据OER过程中关键反应中间体的转化步骤,OER机制中涉及两种主流反应路径:吸附物演变机制(AEM)和晶格氧机制(LOM)。在AEM中,金属位点作为氧化还原中心,涉及多个高度相关的氧中间体,导致其最低过电位的热力学限制为0.37 V。对于LOM,氧位点被激活为氧化还原中心,参与氧的形成,从而突破了AEM的比例关系限制,具有更有利的热力学。然而,主导的LOM路径由于涉及晶格氧以及氧空位的形成和填补,容易导致催化剂的结构不稳定和催化性能下降。毫无疑问,通过同时激活金属和晶格氧位点的氧化还原反应构建AEM-LOM兼容的机制,可以结合每条路径的优点,并调和OER的催化活性和稳定性。然而,在当前以单一组分和配位环境为特征的催化系统中,电子转移过程要么发生在金属位点,要么发生在晶格氧位点,这取决于金属和氧带相对于费米能级的位置。因此,开发一个能够同时激活金属和晶格氧氧化还原对的耦合催化系统仍面临重大挑战。
过渡金属羟基氧化物(MOOH,M = Fe、Co或Ni)是在氧气析出反应(OER)电氧化条件下不可逆结构重构的产物,普遍被认为是真正的活性物质,其中基于NiFe的羟基氧化物正逐渐取代商业化的RuO2和IrO2,成为OER催化剂的基准。特别是,一些近期报告发现,OER重构衍生的高价金属中心的羟基氧化物具有触发晶格氧机制(LOM)以提高OER活性的潜力,这为通过电催化重构策略构建具有合适OER路径的催化剂提供了重要思路。然而,催化剂的电催化结构重构通常仅限于近表面纳米尺度,导致活性组分的利用率较低。NiMoO4水合物具有由四个边共享的NiO6八面体与MoO4四面体连接而成的三维网络,并通过阳极电位驱动的结晶水和Mo的共同浸出展现出良好的结构自重构特性,因此可以被选作适合的自牺牲前催化剂,以构建具有灵活配位结构的氧羟化物。
本文要点
1. 在这项研究中,NiMoO4水合物被用作预催化剂,同时通过化学蚀刻共同引入Fe和S作为调节剂,诱导产生丰富的结构缺陷,并促进在电化学激活过程中完全重构为R-NiFeOOH@SO4活性物质。
2. 表征证据表明,阳极激活触发了金属和晶格氧位点的氧化还原过程,并涉及氧空位的形成和填补。
3. 此外,通过原位衰减全反射傅里叶变换红外光谱(ATR FTIR)和18O同位素标记差分电化学质谱(DEMS),可以确认AEM-LOM兼容的OER催化机制,其中R-NiFeOOH@SO4在Fe和S物种的共同调节下,赋予了同时优化的AEM和LOM路径。
4.密度泛函理论(DFT)计算表明,引入的Fe作为AEM路径的活性位点优化了OER中间体的吸附,而引入的S显著刺激了晶格氧的活性,并增加了OER的LOM路径占有率。在Fe和S的共同调节下,R-NiFeOOH@SO4系统实现了AEM和LOM路径的协同催化,最大限度地利用了表面金属和氧活性位点,提升了OER的催化活性。
图文介绍

图1.催化剂的制备。a,纳米珊瑚状 NiMoO4.xH2O@Fe,S 的合成路线,插图显示了纳米棒表面结构的演变。图 b、f 为NiMoO4.xH2O,c、g 为 NiMoO4.xH2O@Fe,S,d、e、h 为 R-NiFeOOH@SO4 的 FESEM、TEM 和高分辨率 TEM 图像。图 i 为 R-NiFeOOH@SO4 的 HAADF-STEM 图像及相应的 Ni、Mo、O、Fe、S 的 EDS 元素分布。
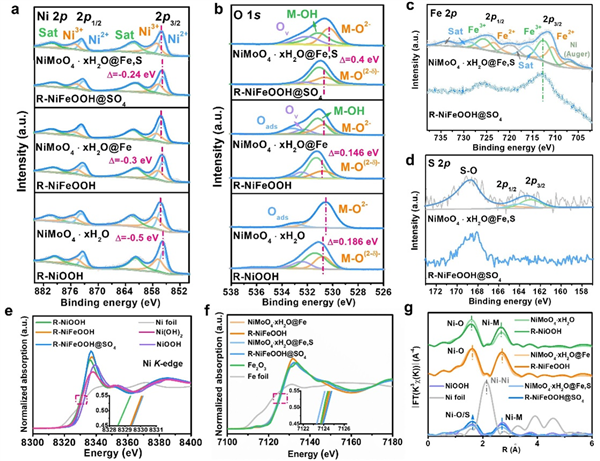
图2. 催化剂电子和配位结构表征。高分辨率 XPS 结果显示了 (a) Ni 2p、(b) O 1s、(c) Fe 2p 和 (d) S 2p 的峰值,比较了获得的样品在 CV 活化前后的变化。(e) 是获得样品和选定参考材料的归一化 Ni K 边和 (f) Fe K 边 XANES 光谱。(g) 是获得样品在 CV 活化前后 Ni K 边光谱的傅里叶变换 k3χ(R) 图。
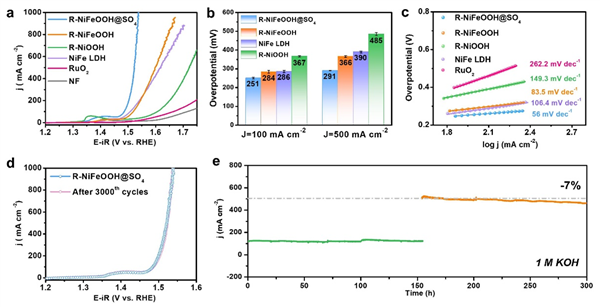
图3. 催化剂OER性能表征。(a) OER 极化曲线,进行了 80% 的 iR 补偿(电极工作面积为 0.5 cm × 0.5 cm,补偿所用的溶液电阻约为 2.3 ± 0.2 Ω);(b) 催化剂在 100 和 500 mA cm-2 下的相应过电位;(c) 催化剂的 Tafel 图;(d) 3000 次 CV 循环前后的 LSV 曲线;(e) R-NiFeOOH@SO4 在 1 M KOH 中于 0.65 和 0.85 V(vs Hg/HgO)下的无 iR 补偿的计时安培曲线。
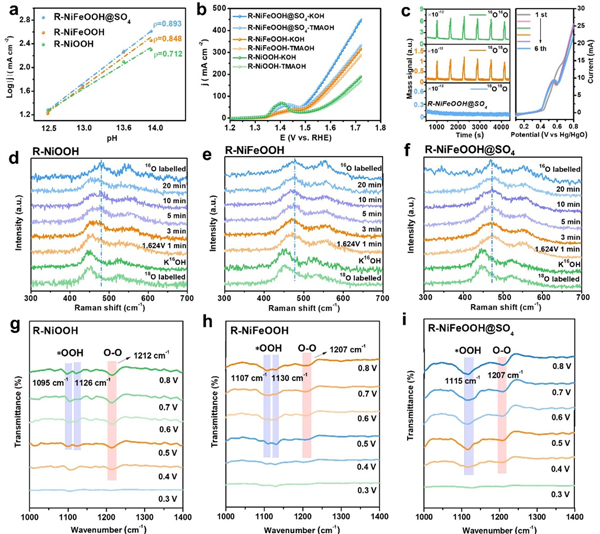
图 4:AEM-LOM OER 催化分析。 (a) 催化剂在相对于 RHE 的 1.7 V 电位下的电流密度对数与 pH 的关系。 (b) 催化剂在 1 M KOH 和 1 M TMAOH 中的 OER 极化曲线,均未进行 iR 补偿。 (c) R-NiFeOOH@SO4 的 DEMS 信号相对于时间的检测结果,未对 16O16O、16O18O 和 18O18O 进行任何校正或减法处理,及相应的 LSV 曲线,未进行 iR 补偿。(d) 18O 标记的 R-NiOOH、(e) R-NiFeOOH 和 (f) R-NiFeOOH@SO4 在 0.1 M KOH 中于 1.624 V 相对于 RHE 测得的原位拉曼光谱。(g) R-NiOOH、(h) R-NiFeOOH 和 (i) R-NiFeOOH@SO4 的原位 ATR-FTIR 光谱。
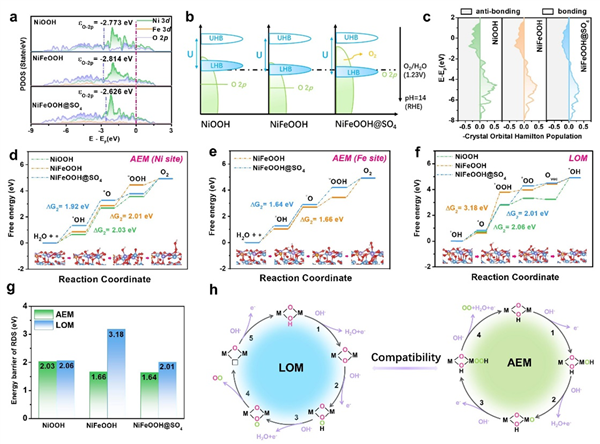
图5. DFT理论计算。(a) 态密度, (b) 示意能带图(UHB 为上 Hubbard 带,LHB 为下 Hubbard 带), (c) Ni-O 键的晶体轨道COHP。 (d) Ni 位点和 (e) Fe 位点的 AEM 途径的 Gibbs 自由能图,以及 (f) LOM 途径的 Gibbs 自由能图。(g) AEM 和 LOM 反应的速率决定步骤(RDS)的能量障碍。
本文信息
Luo, X., Zhao, H., Tan, X. et al. Fe-S dually modulated adsorbate evolution and lattice oxygen compatible mechanism for water oxidation. Nat. Commun. 15, 8293 (2024).
DOI: 10.1038/s41467-024-52682-y
https://www.nature.com/articles/s41467-024-52682-y