第一作者:陈钉
通讯作者:木士春教授
通讯单位:武汉理工大学材料复合新技术国家重点实验室
论文DOI:10.1002/anie.202407577
全文速览
本文采用高温熔盐辅助策略制备了一种由轻质硼(B)原子有序填充的金属间Ir-B化合物(IrB1.1)。位于铱(Ir)晶格中的B通过供-受体结构形成了良好的吸附表面,在析氧反应(OER)的速率决定步骤(RDS)中具有最优的自由能,从而提高了催化活性;同时,Ir-B结构单元的强耦合抑制了Ir的脱金属和重构行为,保证了催化稳定性。这种B诱导的间隙效应使IrB1.1具有比商业IrO2更高的OER性能,并实现了质子交换膜水电解槽(PEMWE)的长期稳定运行。
背景介绍
析氧反应(OER)是制约整个水电解装置效率的关键因素。在酸性介质中,OER催化剂很难在连续氧化过程中保持长期稳定。因此,开发高性价比的Ir基OER催化剂是促进PEMWE广泛应用的关键。然而,目前所设计的掺杂型Ir基催化剂,其位点往往与催化剂宿主晶格的相互作用较弱,易于发生溶解和表面重构。构建有序轻原子填充的金属间化合物将会形成强化学键的结构单元,从而有效抑制高活性金属位点在恶劣工作环境中的降解或失活行为。鉴于此,本文采用高温熔盐辅助策略,实现了B原子在Ir晶格中的有序填充,揭示的间隙效应对设计和构筑具有平衡活性-稳定性的电催化剂提供了科学指导。
本文亮点
1. 利用高温熔盐辅助策略触发轻质B原子在紧密排列的Ir晶格中的有序填充,获得了具有独特“网络”结构的金属间化合物IrB1.1。
2. 揭示了由B诱导的间隙效应。增强活性: 驻留在Ir间隙晶格中的B形成供体-受体结构,调节Ir的电子态和配位环境,从而优化OER中间体的吸附,降低速率决定步骤(RDS)的热力学势垒;保证稳定性: 这种有序填充形成的周期性结构加强了Ir-B结构单元的化学键,并通过强主-客体电子耦合限制了OER过程中Ir活性位的溶解和重建。
3. 获得的IrB1.1实现了OER活性和稳定性上的综合提高,性能表现优于商用IrO2。以IrB1.1作为阳极电催化剂的高性能PEMWE可长期稳定运行。
图文解析
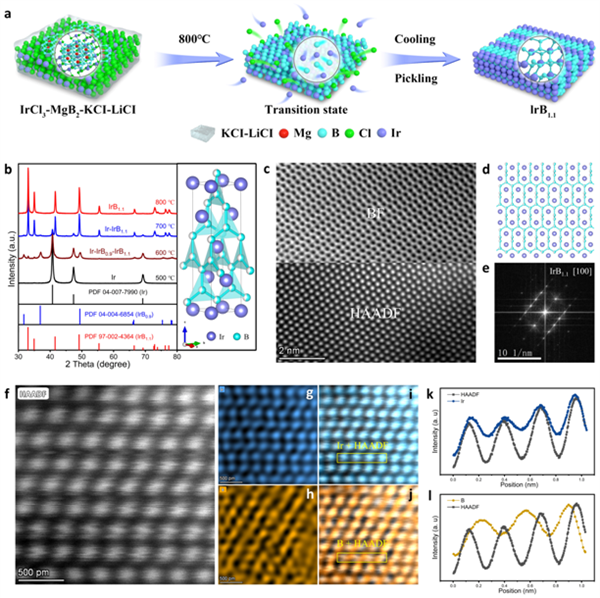
图1 IrB1.1的合成与结构表征
采用高温熔盐辅助策略形成有序的IrB1.1。通过观测在特定区域的亮场(BF)和高角度环形暗场(HADDF),该金属间化合物可匹配[100]取向的IrB1.1投影晶体结构。此外,高分辨率原子图像(图1f)和相应的能量色散X射线谱(EDS)作图结果(图1g-j)证明了B原子在Ir晶格中的有序填充,其中B占据间隙位置。图1i和图1j黄框区域对应的线扫描剖面(图1k、图1l)也从线性分布上揭示了这一点。

图2 IrB1.1的电子和配位结构调查
在高分辨率B 1s光谱中(图2a),186.8和188.2 eV处的两个独立峰分别对应IrB1.1中的B-Ir和B-B键,表明金属间Ir-B硼化物的形成。此外,通过比较Ir 4f光谱中的核心能级Ir 4f5/2和Ir 4f7/2(图2b)可以看出,引入B后,IrB1.1的结合能相对于金属Ir发生了负位移(~0.3 eV)。这表明引入的B原子通过电负性的差异与Ir形成了供-受体结构,为Ir原子创造了一个富电子的环境。这种电子富集可在Ir L3边缘的x射线吸收近边结构(XANES)光谱中得到进一步证明(图2c)。扩展x射线吸收精细结构(EXAFS)光谱(图2e)显示,IrB1.1的主峰归属于Ir-B键引起的Ir和B的散射相互作用。空间拟合结果(图2f)进一步表明,IrB1.1具有两个不同距离的Ir-B路径以及Ir-Ir路径。Ir-B配位的复杂性有利于调节Ir原子的电子结构和反应中间体的吸附能力。
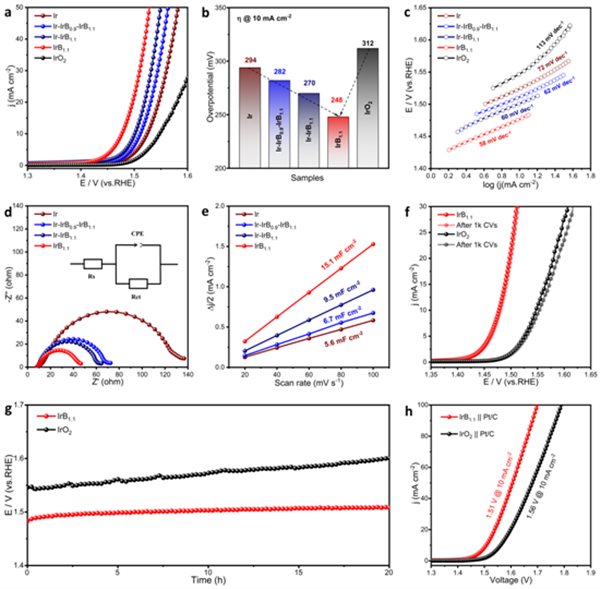
图3 IrB1.1的OER性能研究
极化曲线表明,IrB1.1具有良好的OER活性(248 mV @10 mA cm-2)。Tafel图和Nyquist图显示,与对照样品相比,IrB1.1电荷转移电阻(Rct)降低,说明B的参与提高了IrB1.1的OER动力学过程。在OER稳定性方面,IrB1.1和商业IrO2在1000 圈CV循环前后的极化曲线和计时电流曲线表明,IrB1.1比商业IrO2具有更好的加速降解稳定性和长期循环耐久性。

图4 PEMWES性能研究
将双极板、多孔传输层(PTL)及膜电极等组装成PEMWE,对IrB1.1和IrO2的OER性能进行评估。PTL和压敏纸的表面应力分布云图非常均匀,说明在此压力下(~2.5 Mpa),催化层与PTL有良好的接触,有利于获得优异的性能。在80℃下,PEMWE的极化曲线验证了相比于商业IrO2,IrB1.1对阳极OER的催化活性有所提高。此外,在施加1.65 V的槽电压下,所得电解槽提供约500 mA cm-2的电流密度,并可保持长达100 h的水电解活性。这些实验结果证实了金属间化合物IrB1.1在PEMWE中具有光明的应用前景。
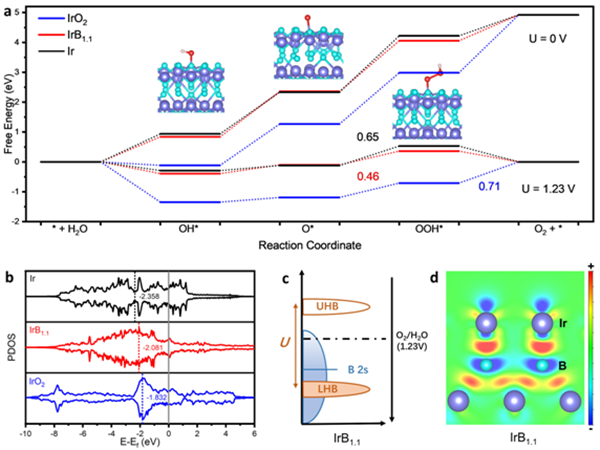
图5 IrB1.1的OER活性起源分析
为了更深入地了解IrB1.1金属间化合物增强的OER性能,进一步构建了包括IrB1.1、IrO2和Ir在内的相关结构模型,并进行了密度泛函理论(DFT)计算。如图5a所示,在1.23 V时,OOH*在IrO2上生成O2是速率决定步骤(RDS),其自由能差为0.71 eV;而对于Ir和IrB1.1的RDS为O*到OOH*的转化。很明显,在Ir晶格中引入间隙B大大降低了RDS的势垒。同时,与IrO2相比,IrB1.1对含氧中间体的吸附减弱,这也解释了为什么IrB1.1具有比IrO2更好的OER活性。
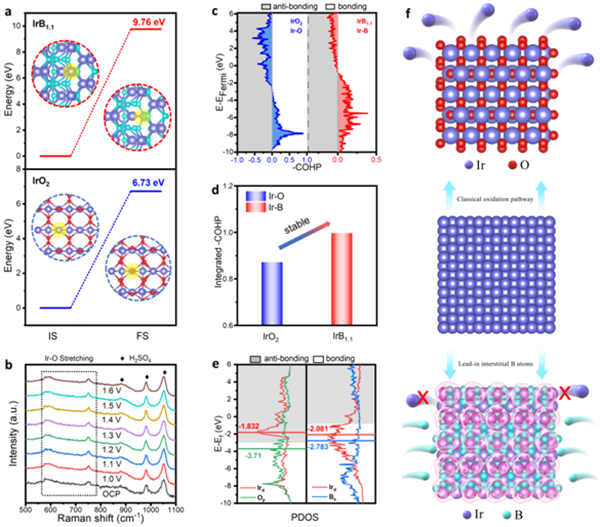
图6 IrB1.1耐久性机制分析
在稳定性方面,首先计算了Ir在IrB1.1和IrO2表面的Ir脱金属能,用以解释催化剂稳定性的差异。这是因为活性原子的溶解或脱失是催化剂稳定性低的重要原因之一。如图6a所示,IrB1.1的脱金属能垒(9.76 eV)高于IrO2的脱金属能垒(6.73 eV),表明活性金属溶解量较少。此外,原位拉曼光谱表明,IrB1.1中Ir-O拉伸保持在相同的位置,没有出现与电位相关的峰,证明IrB1.1在实际OER过程中能够保持良好的结构稳定性。此外,通过计算COHP来进一步评估催化剂的化学键强度。计算结果表明,IrB1.1中形成了更强的化学键。因此,B诱导的间隙效应加强了结构单元之间的化学键,并且Ir-B的强电子耦合抑制了Ir的溶解和重构,从而增强了OER稳定性。
总结与展望
综上所述,本文设计并构建了有序填充B原子的金属间化合物IrB1.1作为酸性OER催化剂,并揭示了B诱导的间隙效应对催化活性和稳定性的提升和促进作用。引入的B与宿主Ir原子形成供体-受体结构,调节Ir的电子态和配位环境,从而优化OER中间体的吸附,降低速率决定步骤的热力学势垒。此外,这种有序填充形成的周期性结构限制了Ir的溶解或重构。因此,使用IrB1.1催化剂实现了酸性介质中OER活性和稳定性的综合提高,并实现了PEMWE的长期稳定运行。这项工作揭示的间隙效应加深了对金属间化合物催化机制的理解,并将激励人们加大对该类型催化剂的开发力度。
作者介绍
陈钉,武汉理工大学木士春教授课题组2021级博士研究生,主要研究方向为催化剂的设计合成与催化机制。目前以第一作者在Nat. Commun.、Angew. Chem. Int. Ed.、Energy Environ. Sci.、Adv. Energy Mater.、ACS Energy Lett.、ACS Catal.、Nano Energy、InfoMat、Appl. Catal. B: Environ.等期刊上发表10余篇高质量学术论文。
木士春,武汉理工大学首席教授,博士生导师,国家级高层次人才。长期致力于质子交换膜燃电池和电解水催化剂研究。以第一作者或通讯作者在Nat. Commun.、Adv. Mater.、J. Am. Chem. Soc.、Angew. Chem. Int. Ed.、Energy Environ. Sci.、Nano Lett.等国内外期刊上发表300余篇高质量学术论文。
木士春研究团队主页http://shichunmu.polymer.cn/