- 学科首席教授!武汉理工大学「国家级高层次人才」木士春,新发Nature子刊
- 来源:木士春教授研究团队 个人网站 2024-10-04
科研动态 生化环材圈 2024年09月29日 11:00 上海
同时激活金属和晶格氧位点以构建兼容的多机理催化,有望为析氧反应(OER)提供高可用性的活性位点,并提高催化活性/稳定性,但这仍是一项重大挑战。
2024年9月27日,武汉理工大学木士春教授团队在Nature Communications期刊发表题为“Fe-S dually modulated adsorbate evolution and lattice oxygen compatible mechanism for water oxidation”的研究论文,团队成员罗旭为论文第一作者,木士春教授为论文通讯作者。
该研究通过在OER过程中对NiMoO4·xH2O@Fe,S进行完全重构,获得了Fe和S双调制的NiFe氢氧化物(R-NiFeOOH@SO4),并通过同时优化金属/氧位点实现了兼容的吸附质演化机制和晶格氧氧化机制,这一点已通过原位光谱/质谱分析和化学探针得到证实。进一步的理论分析表明,Fe促进了吸附质演化机理下的OER动力学,而S激发了晶格氧氧化机理下的晶格氧活性,表现为O 2p带中心上移、d-d库仑相互作用扩大、金属-氧键减弱、中间吸附自由能优化。得益于兼容的多机理,R-NiFeOOH@SO4只需要251±5/291±1 mV的过电位就可以在碱性介质中驱动100/500 mA cm-2的电流密度,并且能稳定运行超过300小时。该研究有助于深入理解OER机理,从而更好地设计高性能OER催化剂。
该研究采用水合物NiMoO4作为预催化剂,同时通过化学刻蚀法引入Fe和S作为调制剂,从而在电化学活化过程中诱发大量结构缺陷,并促进R-NiFeOOH@SO4活性物质的完全重构。表征结果表明,阳极活化触发了金属和晶格氧位点的氧化还原,并涉及氧空位的形成和再填充。此外,通过原位衰减全反射傅立叶变换红外光谱(ATR FTIR)和18O同位素标记差分电化学质谱(DEMS)证实了AEM-LOM相容的OER催化机理,其中R-NiFeOOH@SO4在Fe和S两种物质的协同调节下同时具有最优的AEM和LOM反应途径。密度泛函理论DFT计算表明,引入Fe作为AEM途径的活性位点,优化了OER中间体吸附,而引入S显著刺激了晶格氧活性,增加了OER的LOM反应途径的占用率。通过Fe和S的协同调节,R-NiFeOOH@SO4体系实现了AEM和LOM途径的协同催化,最大限度地利用了表面金属和氧活性位点,提高了OER催化活性。

图1. a纳米珊瑚样NiMoO4.xH2O@Fe,S的合成路线,插图显示了纳米棒的表面结构演变的放大图像。b, f NiMoO4.xH2O、c, g NiMoO4.xH2O@Fe,S和d, e, h R-NiFeOOH@SO4的FESEM、TEM和高分辨率TEM图像。(插图为放大图,图g中为相应选区的电子衍射图)。i R-NiFeOOH@SO4的HAADF-STEM图像及其对应的Ni, Mo, O, Fe, S的EDS元素映射图。
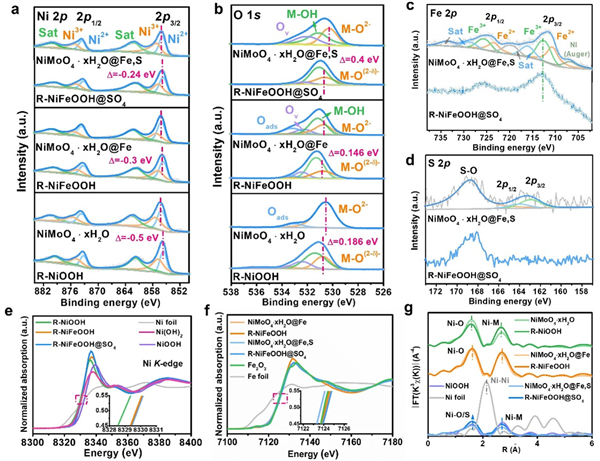
图2. a-d获得的的样品在CV活化前后的a Ni 2p、b O 1s、c Fe 2p和d S 2p的高分辨率XPS光谱。e、f 所得到的样品和选定参考物质的e 归一化Ni K-edge、f Fe K-edge XANES光谱。插图显示了所选区域的放大视图,x轴:能量(eV), y轴:归一化吸收(a.u)。g CV活化前后样品的傅立叶变换k3χ(R) Ni K-edge谱。
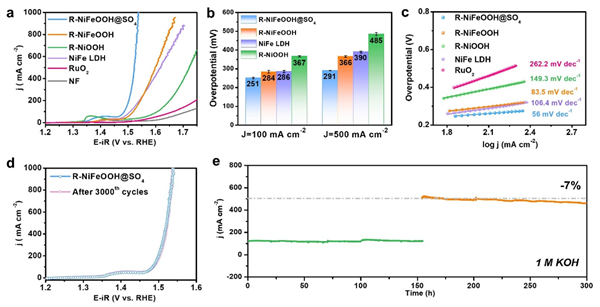
图3. a 80% iR补偿后的OER极化曲线(电极工作面积为0.5 cm × 0.5 cm,用于补偿的溶液电阻约为2.3±0.2 Ω),b 在100和500 mA cm-2下催化剂的相应过电位。c 催化剂的塔菲尔图。d 3000 CV循环前后的LSV曲线。e 在0.65和0.85 V下,1 M KOH中不含iR补偿R-NiFeOOH@SO4的计时电流曲线。

图4. a 1.7 VRHE电位下催化剂的电流密度对数和pH之间的关系。b 1 M KOH和1 M TMAOH中不含iR补偿的催化剂的OER极化曲线。c检测到的DEM信号,对R-NiFeOOH@SO4的16O16O、16O18O、18O18O相对于时间和相应的LSV曲线不做任何校正或省略,不做iR补偿。d-f 分别为在1.624 VRHE下,在0.1 M KOH和H216O中测量的18O标记的d R-NiOOH、e R-NiFeOOH和f R-NiFeOOH@SO4的原位拉曼光谱。16O标记样品的拉曼光谱放在顶部进行比较分析。g-i 分别为g R-NiOOH、h R-NiFeOOH、i R-NiFeOOH@SO4的原位ATR-FTIR光谱。

图5. a 投影态密度。b 能带示意图(UHB上哈伯德带,LHB下哈伯德带)和c Ni-O键的晶体轨道汉密尔顿居群(COHP)。d-f 分别为AEM途径在d Ni位点和e Fe位点上的吉布斯自由能图;以及f LOM途径上Ni位点和Fe位点上的吉布斯自由能图。g AEM和LOM的RDS能垒。以上数据基于NiOOH、NiFeOOH和NiFeOOH- SO4。h NiFeOOH@SO4上AEM和LOM途径示意图。
总之,该研究设计了Fe、S物种协同调节的NiMoO4水合物(NiMoO4.xH2O@Fe,S)作为预催化剂,调节NiMoO4.xH2O的晶体和电子结构,促进其完全重构为R-NiFeOOH@SO4活性物质。XPS和XAFS分析表明,阳极活化同时触发金属和晶格氧位点的氧化还原。电化学和化学探针分析表明,与衍生的R-NiOOH和R-NiFeOOH相比,S物种修饰的R-NiFeOOH@SO4具有更高的晶格氧活性和更有利的LOM反应倾向。原位18O同位素标记DEM和ATR FTIR光谱证实了R-NiFeOOH@SO4的AEM-LOM兼容OER机理,并在Fe和S的共调制下同时优化了AEM和LOM反应途径。DFT计算分析进一步揭示了Fe和S在调节反应中的关键作用,并阐明了OER活性提高的原因。Fe通过其活性位点优化了AEM途径下OER中间体的吸附自由能,而S则提高了LOM途径下RDS的晶格氧活性,降低了RDS的能垒,通过提高AEM和LOM途径的兼容性共同优化了OER活性。因此,R-NiFeOOH@SO4活性衍生物在碱性介质中对OER具有较高的本征活性和稳定性。该研究为通过合理激发晶格氧和金属氧协同位点设计高稳定、高活性的非贵金属催化剂提供了启示。
- [来源:中国聚合物网]
- 了解更多请进入: 木士春教授研究团队 个人网站